ABSTRACT
We present the case of a 53-year-old woman of Portuguese ancestry with a diagnosis of progressive systemic sclerosis (SSc), proposed for haematopoietic stem cell transplantation (HSCT). Clinical re-evaluation when assessing eligibility for the procedure led to the alternative diagnosis of familial amyloid polyneuropathy (FAP). We discuss the clinical presentations of FAP and SSc, focusing on their overlapping and distinguishing features. We emphasize the need for a high level of suspicion in order to establish an early diagnosis of FAP in the absence of a family history, and provide prognostic and genetic counselling.
LEARNING POINTS
- It is important to review diagnoses, especially when the clinical course is atypical.
- Cutaneous involvement is a commonly unrecognized feature of familial amyloid polyneuropathy.
- Hereditary conditions should be included in the differential diagnosis of multisystemic diseases, even in the absence of a family history.
KEYWORDS
Hereditary, amyloidosis, transthyretin, scleroderma, polyneuropathy
CASE DESCRIPTIONS
A 53-year-old woman, who had retired due to disability, was referred by her rheumatologist to our centre for haematopoietic stem cell transplantation (HSCT), for progressive systemic sclerosis with severe multisystemic involvement, which was unresponsive to several lines of treatment.
Her medical history included carpal tunnel syndrome, restless leg syndrome, neurogenic bladder, hypothyroidism and third-degree atrioventricular block requiring a pacemaker at the age of 49, diagnosed in this order. Systemic sclerosis (SSc) had been the last diagnosis added to the list, based on the findings of generalized skin thickening, extremity swelling, Raynaud phenomenon, chronic kidney disease and severe gastrointestinal dysmotility including constipation followed by diarrhoea, pyrosis and dysphagia. Her medication included cyclophosphamide, sildenafil, pentoxifylline, levothyroxine, ramipril and prokinetics. Immunosuppressants such as mycophenolate mofetil and azathioprine had also been used unsuccessfully. The degree of dysmotility was so severe that the patient had undergone percutaneous endoscopic gastrostomy.
At re-evaluation, vital signs were unremarkable except for low systolic pressure. Sclerodactyly and limited mouth opening due to deep skin thickening was confirmed. Dupuytren's contracture of the fifth finger of both hands, petechiae and bruises were also evident. Although no digital ulcers were found, leg blisters and scars at various stages of evolution were seen (Fig. 1). Neurological examination revealed symmetric loss of thermoalgic sensitivity in the lower and upper limbs (up to the knees and elbows) which had not been previously described. When asked, the patient confirmed to have noticed neuropathic pain for the last 10 years and progressive loss of sensibility for water temperature for at least 7 years, which resulted in successive burns.
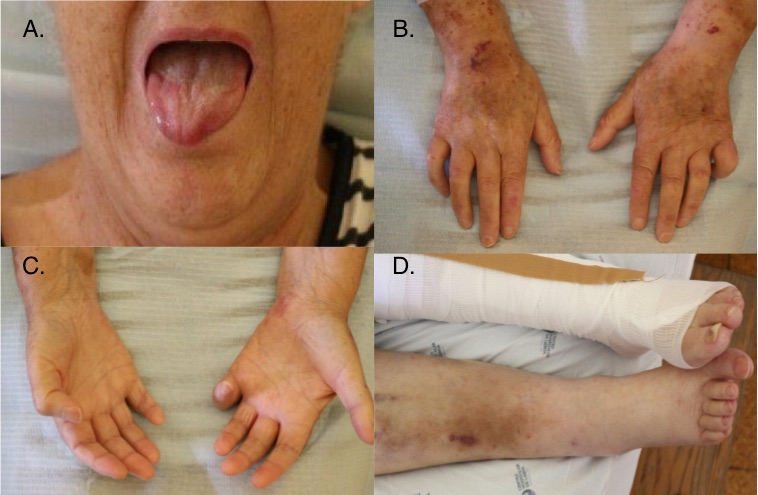
Figure 1 (click to enlarge)
Figure 1. (A) Limited mouth opening; (B,C) sclerodactyly, signs of vascular fragility and Dupuytren's contractures; (D) lower limb oedema, scars and hyperpigmentation
Ascendent and symmetrical peripheral neuropathy together with signs of vascular fragility, gastrointestinal dysmotility and cardiac conduction abnormalities raised suspicion of systemic amyloidosis. The family history revealed two daughters, 35 and 37 years old, with bilateral severe carpal tunnel syndrome. There was also a record of alcoholic liver cirrhosis in the patient’s father and brother, which had been the presumptive cause of their premature deaths in their fifties. It was found that the patient originated from a region where a small hereditary amyloidosis cluster was known. Diagnostic evaluation was conducted accordingly.
Methods and Procedures
Diagnostic tests revealed normocytic anaemia (haemoglobin 9.7 g/dl) with an increased erythrocyte sedimentation rate of 66 mm/hr, with no other inflammatory markers; liver parameters were normal; NT-pro-BNP was 280 pg/ml; chronic renal disease was exacerbated (urea 50.71 mmol/l, creatinine 247.58 μmol/l) with non-nephrotic proteinuria; antinuclear and scleroderma-related autoantibodies were negative. Nailfold capillaroscopy showed minor dysmorphisms, without a scleroderma pattern, contrasting with the advanced stage of the disease. Transthoracic echocardiography revealed bi-ventricular and septal wall thickening with diastolic dysfunction. Chest radiography and lung function tests were normal. Upper digestive tract endoscopy showed mild gastritis with preserved wall distention.
Oesophageal manometry revealed the absence of primary peristalsis of the medial and distal portions of the oesophagus. Electromyography (EMG) showed axonal polyneuropathy, with sensory involvement greater than motor, and chronic denervation and reinnervation. Abdominal fat pad biopsy exhibited amyloid deposition positive to anti-transthyretin antibodies. Primary and secondary amyloidosis were excluded. Genetic testing confirmed familial amyloid polyneuropathy, with mutation of the transthyretin gene (Val30Met).
Unfortunately, the patient was not eligible for targeted treatment due to the advanced state of the disease and ended up dying from complications at the age of 56. However, her daughters were promptly referred to a geneticist and started follow-up at a reference centre as the same diagnosis was confirmed.
DISCUSSION
Familial amyloid polyneuropathy (FAP), or transthyretin-related amyloidosis (ATTR), is an autosomal dominant hereditary form of systemic amyloidosis first described by neurologist Corino de Andrade in Portugal [1]. For some time, it was considered an endemic disease. Later the same variant was found in Sweden, Brazil and Japan, possibly linked by commercial trade between these countries. Many independent mutations have since been identified all over the world.
It is a multisystemic infiltrative, degenerative disease, involving irreversible deposition of misfolded amyloidogenic variants of TTR in the peripheral nervous system and organs such as the heart, kidney and skin. First symptoms usually manifest between the fourth and fifth decades of life and include sensorimotor polyneuropathy with neuropathic pain, loss of thermoalgic sensitivity of the extremities, and carpal tunnel syndrome. These are considered hallmarks, together with dysautonomia, explaining gastrointestinal tract and bladder dysmotility. It is common for patients to develop bradyarrhythmias requiring a pacemaker due to the cardiac conduction system being affected by septal deposition of amyloid. Although skin deposits can be histologically identified early in the course of the disease, its clinical manifestations usually only become evident in the terminal stages, and include alopecia, oedema, epidermal fragility and dermal thickening, similar to scleroderma. Raynaud phenomenon, hypothyroidism, xerostomia and xerophthalmia can also occur, mimicking autoimmune disorders [2].
Systemic sclerosis (SSc) is a rare pleomorphic disease characterized by autoimmunity, vasculopathy and fibrosis. Stimulated fibroblasts prompt the deposition of collagen and other extracellular proteins in the dermis and internal organs, leading to skin thickening, sclerodactyly, Raynaud phenomenon, digital ulcers, telangiectasia and contractures. Multisystemic involvement can comprise gastrointestinal tract dysmotility with dysphagia, pulmonary fibrosis, and heart and kidney failure. Diagnosis is clinical, but laboratory tests like abnormal nailfold capillaroscopy and scleroderma-related autoantibodies can favour the diagnosis, which can be very challenging when these are absent. There is, so far, no specific treatment. Several immunosuppressors have been tried and HSCT is indicated in selected patients with rapidly progressive SSc at risk of organ failure. Although our patient apparently met the 2013 European classification criteria for systemic sclerosis, an important nuance in such criteria is the fact they are not applicable to patients having a disorder better explaining their symptoms [3]. Likewise, severe peripheral polyneuropathy is not a common feature of this disease.
In the presence of such findings, EMG should be requested. Findings in FAP include axonal polyneuropathy, with sensory involvement greater than motor, and evidence of chronic denervation and reinnervation. Tissue biopsy is essential, showing amyloid deposition by Congo red staining and positive immunolabeling with anti-TTR antibodies. Diagnosis is confirmed by genetic testing, identifying mutations in the TTR gene.
Since the liver is the main site of production of TTR, disease stability can be achieved with liver transplantation. Two drugs have also been shown to delay disease progression, tafamidis and patisiran. Early diagnosis is essential as treatment amenability and prognosis depend on the extent of target organ damage, specially of the heart and kidneys.
This case reveals the challenges in diagnosing complex and rare diseases with multisystemic involvement. It highlights the importance of a thorough clinical evaluation and clinical history, and demonstrates that cutaneous involvement is a commonly unrecognized feature of FAP[4]. Hereditary conditions are frequently overlooked and should always be considered in the differential diagnosis of multisystemic disorders, even in the absence of a family history.