ABSTRACT
Rosai–Dorfman disease (RDD) is a rare disorder characterized by the proliferation of histiocytes in lymph nodes. It can occur anywhere in the body but commonly involves the cervical area of the neck. Its clinical presentation varies and patients with skin manifestations may develop papules, nodules, plaques, or pustules. Histologically, it typically presents with emperipolesis, where intact lymphocytes are found within histiocytes. The definitive treatment of RDD is not well established given the rarity of the disease and indeed skin lesions can regress spontaneously. Therapeutic treatment options include cryotherapy, radiation, or topical agents such as steroids or retinoids. Here we describe the case of a 24-year-old Hispanic female who presented with skin manifestations which proved to be histologically positive for Rosai–Dorfman disease. The patient clinically improved following the administration of intralesional steroids.
LEARNING POINTS
- RDD is a rare self-limiting benign lymphoproliferative disorder, which can be associated with autoimmune disease, hematological malignancies, and post-infectious conditions.
- Skin lesions can be self-limiting in many cases of RDD.
- Therapeutic treatment options include cryotherapy, local radiation, and topical steroids and retinoids.
KEYWORDS
Rosai–Dorfman disease, cutaneous type, lymphoproliferative disorder, emperipolesis
CASE DESCRIPTION
A 24-year-old Hispanic female presented with an acute non-pruritic painless, violaceous skin lesion on her right upper back. The patient had noticed the lesion nine months previously, which had progressively increased in size despite treatment with topical steroids and systemic antifungal medications. The patient had no additional skin lesions, fever, weight loss, recent change to her topical or systemic medications, or recent trauma to the affected area. The patient was afebrile on presentation. On physical examination, an irregular and painless erythematous area, measuring 5 × 7 cm, of multiple papules, nodules, and plaques with clear margins was noted over her right scapula but with no cervical or axillary lymphadenopathy (Fig.1). Autoimmune disease, such as cutaneous lupus erythematosus tumidus, was initially high on our list of differential diagnoses. However, autoimmune analysis, including antibodies for ANA, anti-dsDNA, anti-Jo-1, anticardiolipin, anticentromere-B, antiscleroderma-70, Sjogren’s SSA, SSB, and anti-Smith, were negative. Additionally, the patient’s white blood cell count was within normal limits (4.4 × 103 µL) as was her hemoglobin (14.6 g/dL).
The patient then underwent a shave biopsy, which was significant for dense diffuse inflammatory cells infiltrated with a collection of neutrophils and histiocytes surrounded by lymphocytes and many plasma cells. The histopathology report suggested granulomatous and focally suppurative dermatitis; however, no infectious microorganisms were detected in sections stained with periodic acid-Schiff with diastase, acid-fast bacillus, fluorescein isothiocyanate (FITC), the immunohistochemical stain for Treponema pallidum, or Steiner stains for spirochetes. Lyme serology IgG/IgM and rapid plasma reagin were also negative.
The lesion was treated as a granulomatous lesion secondary to inflammation and the patient prescribed the topical corticosteroid halobetasol propionate twice daily for a 2-week course. However, upon review of the lesion, there was no clinical improvement. It was decided to conduct a punch biopsy of the medial and lateral aspect of the lesion, which showed extranodal sinus histiocytosis with a definitive diagnosis of Rosai–Dorfman disease. Further work-up was undertaken, which included a positron emission tomography computerized tomography (PET CT) scan to rule out other additional extranodal sites. This was negative for hypermetabolic osteolytic or osteoblastic metastatic skeletal lesions. The lesion was treated with intralesional triamcinolone acetonide injectable suspension 10 mg/mL, with an initial dose of 1 mg followed by 0.5–1 mg per injection site, for a total of >7 injectable site lesions. The intralesional injections were repeated weekly and the patient completed a total of eight sessions. The lesion was responsive to the steroid therapy with minimal granulomatous papules; post-inflammatory hyperpigmentation and dry skin were noted as well (Fig. 2).
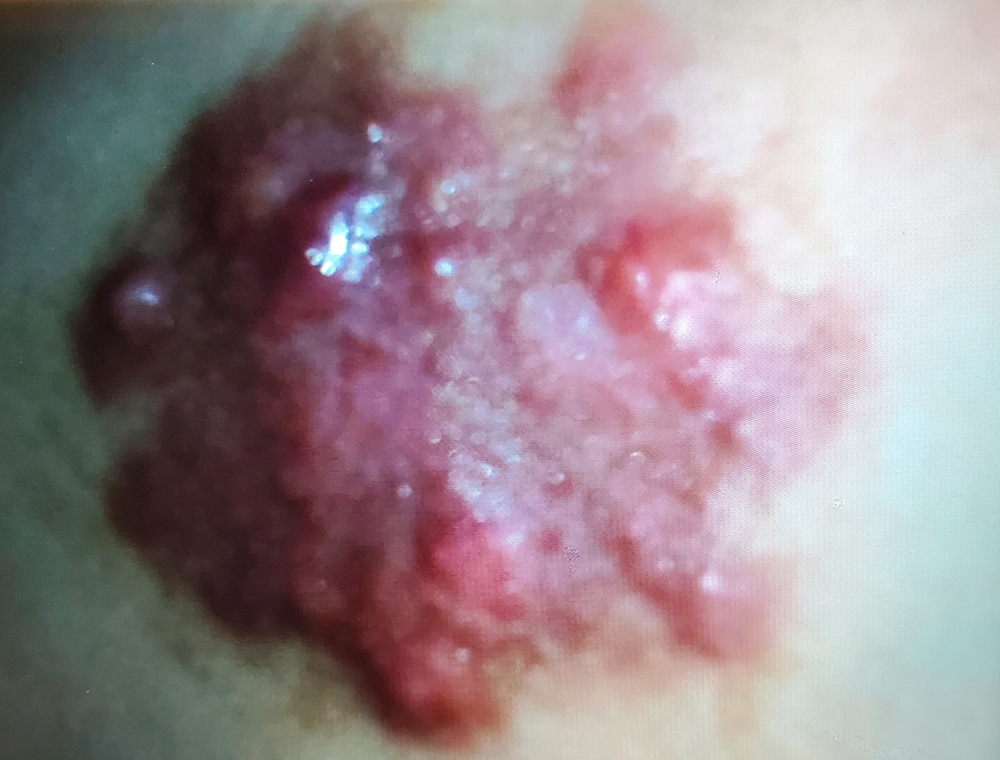
Figure 1 (click to enlarge)
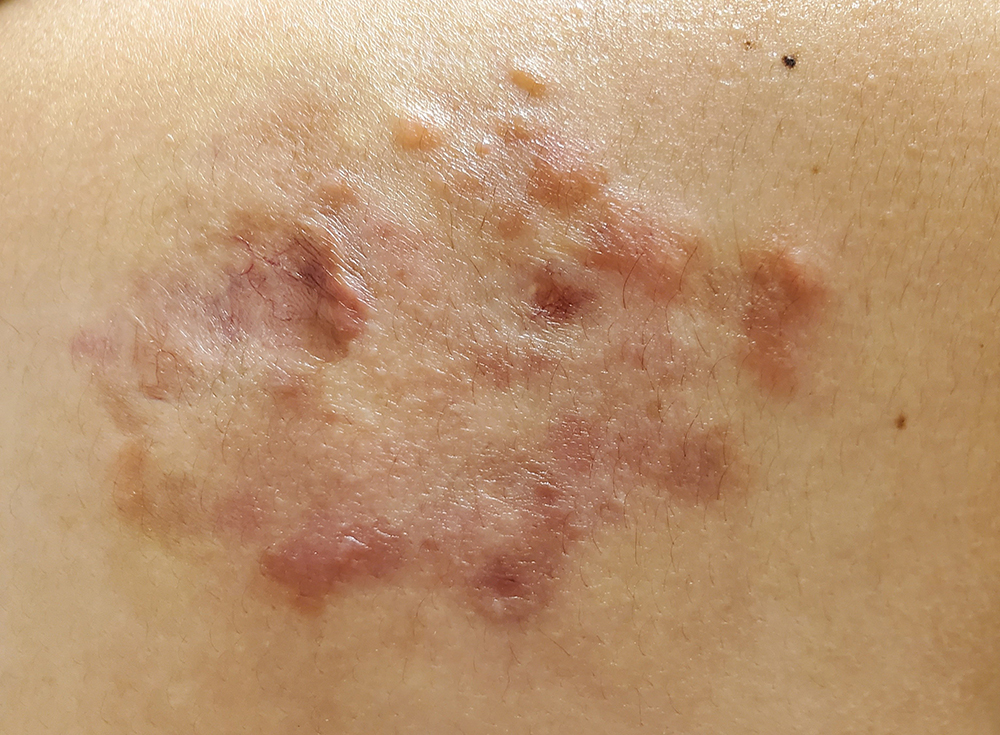
Figure 2 (click to enlarge)
Figure 1. Well-demarcated inflammatory plaques formed by multiple coalescing, yellowish erythematous papules and nodules in the right scapular region
Figure 2. Clinical improvement of the lesion showing few residual granulomatous papules and small plaques after treatment
DISCUSSION
RDD is a rare, often self-limiting, non-malignant lymphoproliferative disorder. It has two main types: the nodal type, in which the cervical lymph nodes are most commonly involved; or the extranodal type, which accounts for 40–43% of cases. The skin (cutaneous RDD) represents the most commonly involved extranodal site (10% of cases), followed by the bones, the nasal cavity and paranasal sinuses, the eyes, the salivary glands, the central nervous system, the oral cavity, the kidneys, the gastrointestinal tract, and the respiratory tract. The disease can also involve the liver, tonsils, breast, and heart[1,2].
Cutaneous RDD usually presents at a later age of onset, with a median age of 43.5 years compared with RDD in general, which has a median age of onset of 20.6 years. Additionally, cutaneous RDD is more common in females (2:1) and usually affects those of Asian and Caucasian ethnicity. Conversely, the other type of RDD is more prevalent in males (1.4:1). Most RDD cases have been reported in patients of African ethnicity, while it is rarely found in Asian patients[2]. Our patient is a Hispanic female who presented with rare skin involvement, with an age younger than the median age of onset.
The pathogenesis of RDD is unknown and multiple hypotheses have been proposed to explain it. The condition has been reported in many patients with autoimmune disease, hematological malignancies, and post-infectious conditions[3]. It is more likely to be a reactive process rather than a neoplastic disorder, given the polyclonal nature of the cell infiltrate and the clinical progression of the disease. Additionally, immune dysfunction and some viral infections were found to be related to its pathogenesis. These viruses include human herpesvirus 6, Epstein–Barr virus, human papillomavirus 6, polyomavirus, cytomegalovirus, and parvovirus B19[1–3].
The histological findings in RDD typically show a diffuse infiltrate of predominantly histiocytes with a background infiltrate of lymphocytes and plasma cells and occasionally lymphoid follicles.
The hallmark of cutaneous RDD is the phenomenon of emperipolesis, or nondestructive phagocytosis, which is determined by the presence of intact lymphocytes within histiocytes[3]. In RDD, the histiocytes tend to stain positive for CD68, CD14, fascin, CD163, HLA-DR, and S100; however, they stain negatively for CD1a. These findings can help to establish the diagnosis of RDD[1–3] and rule out other conditions such as Langerhans cell histiocytosis, histiocytic sarcoma, classical Hodgkin lymphoma, melanoma, metastatic carcinomas, and infectious lymphadenopathies[1,4].
The clinical diagnosis of RDD is challenging as the clinical presentation is usually variable, especially the extranodal type in the absence of lymphadenopathy. Cutaneous RDD may present as single or multiple lesions. These lesions are usually papules, nodules, plaques, pustules, or a combination of these lesions. The disease commonly involves the face, followed by the back, chest, thigh, flank and shoulders[2]. In 1978, Thawerani et al. reported the first case of purely cutaneous RDD in a 48-year-old male patient with a solitary nodule on the shoulder[5]. In our case, the patient presented with multiple, non-tender erythematous combinations of papules, nodules, and plaques in the right scapular area. This non-specific clinical picture makes establishing a clinical diagnosis challenging without histopathological confirmation.
The definitive diagnosis of RDD is made by histopathological examination of the biopsied lesion and immunohistochemical staining to help differentiate RDD from other diseases. In certain cases, a repeat biopsy is indicated, as in our clinical presentation (shave and punch), in order to make a proper judgement and definitive diagnosis. Some patients with RDD may also have elevated C-reactive protein, erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia; however, these laboratory results are non-specific[1]. RDD has shown to have a good prognosis, particularly the cutaneous subtype; however, lower respiratory tract or hepatic involvement is considered a poor prognostic factor. The major causes of mortality include amyloidosis or vital organ involvement[4].
There is no standard treatment for RDD given its rare presentation. The skin lesions may regress spontaneously (20–25%) and only require clinical monitoring. Other options include cryotherapy, local radiation, and steroid and retinoid therapy. In refractory cases, dapsone, imatinib, and thalidomide may be used, yet more research is needed to confirm their efficacy[1, 2]. Surgical excision may be indicated in symptomatic lesions with vital organ involvement or extranodal disease that is potentially resectable[4]. Our patient was treated with topical steroids with no improvement; however, the intralesional injection of steroids resulted in considerable clinical improvement.