ABSTRACT
Abernethy malformation is a rare congenital extrahepatic portosystemic shunt with variable clinical manifestations, mainly associated with the portosystemic shunt. Hepatic encephalopathy can be the first manifestation.
We present the case of a 50-year-old woman with hepatic encephalopathy without liver dysfunction. Further evaluation with contrast-enhanced abdominal computed tomography was performed, showing a large-calibre portocaval shunt from the left portal branch, suggestive of type 2 Abernethy malformation. The patient improved with lactulose and rifaximin but maintained rare encephalopathy episodes. She is therefore currently awaiting surgical treatment.
Despite being a rare cause of hepatic encephalopathy, Abernethy malformation should be considered in patients with unexplained hyperammonaemia. Since it is potentially reversible, and early diagnosis and treatment may improve outcome, raising awareness of this malformation is essential.
LEARNING POINTS
- Abernethy malformation is a rare congenital extrahepatic portosystemic shunt with variable clinical manifestations, some associated with the portosystemic shunt; hepatic encephalopathy can be the first manifestation.
- Initial recognition of the presence of encephalopathy in non-cirrhotic individuals can be challenging, and patients may have symptoms for a long period of time before being diagnosed.
- This entity, although rare, should be looked for in patients with hepatic encephalopathy without evidence of liver disease, mainly because it can be submitted to targeted treatment.
KEYWORDS
Abernethy malformation, portosystemic shunt, encephalopathy, hyperammonemia
CASE DESCRIPTION
A 50-year-old woman was sent for internal medicine consultation because of a 2-year history of repeated episodes of attention impairment and disorientation (with varying degrees of severity). She had already been to the emergency department (ED) twice for these episodes. At the second presentation, she was diagnosed with metabolic encephalopathy and discharged after lactulose and fluid therapy.
She had a personal history of peripheral venous insufficiency. She did not take chronic medication and denied alcohol or drug abuse.
On observation, she was alert and oriented, could follow complex commands, and did not have focal neurological deficits. The remainder of the physical examination was unremarkable, without telangiectasias, signs of abdominal collateral circulation, or ascites. No flapping was observed.
Blood samples drawn in the ED showed increased serum ammonia (Table 1). Furthermore, she had a recent abdominal ultrasound and brain magnetic resonance imaging, which were both normal.
At the consultation, laboratory testing was repeated, showing ammonia levels lower than previously but still increased compared with normal values (64 µg/dl). In addition, the serological evaluation was negative for ceruloplasmin, hepatitis viruses (A, B, C and E) and immunological markers. Contrast-enhanced computed tomography (CT) of the abdomen was performed, showing a portocaval shunt from the left portal branch with a diameter of 13 mm suggestive of type II Abernethy malformation (Fig. 1). Ultrasound liver elastography showed normal stiffness.
Therefore, the patient’s symptoms were attributed to hepatic encephalopathy due to portosystemic shunt. The patient began lactulose and rifaximin (a non-absorbable antibiotic) with improvement and reduction in encephalopathy episodes, but not complete resolution. As a result, she was referred to vascular surgery for evaluation for shunt closure and is currently awaiting surgical treatment.
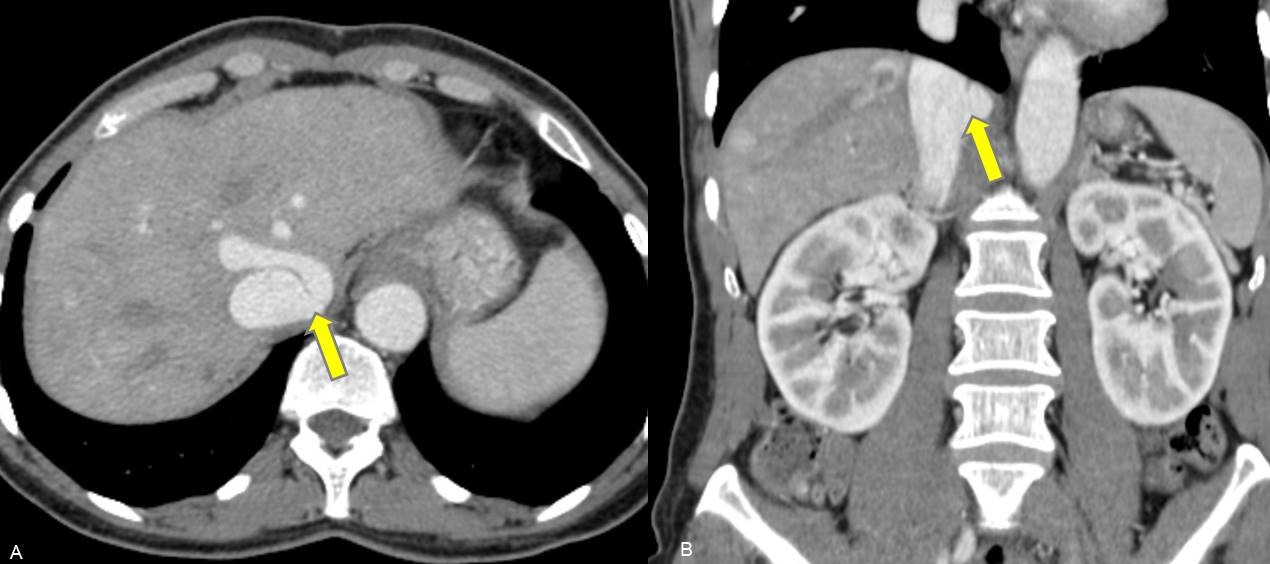
Figure 1 (click to enlarge)
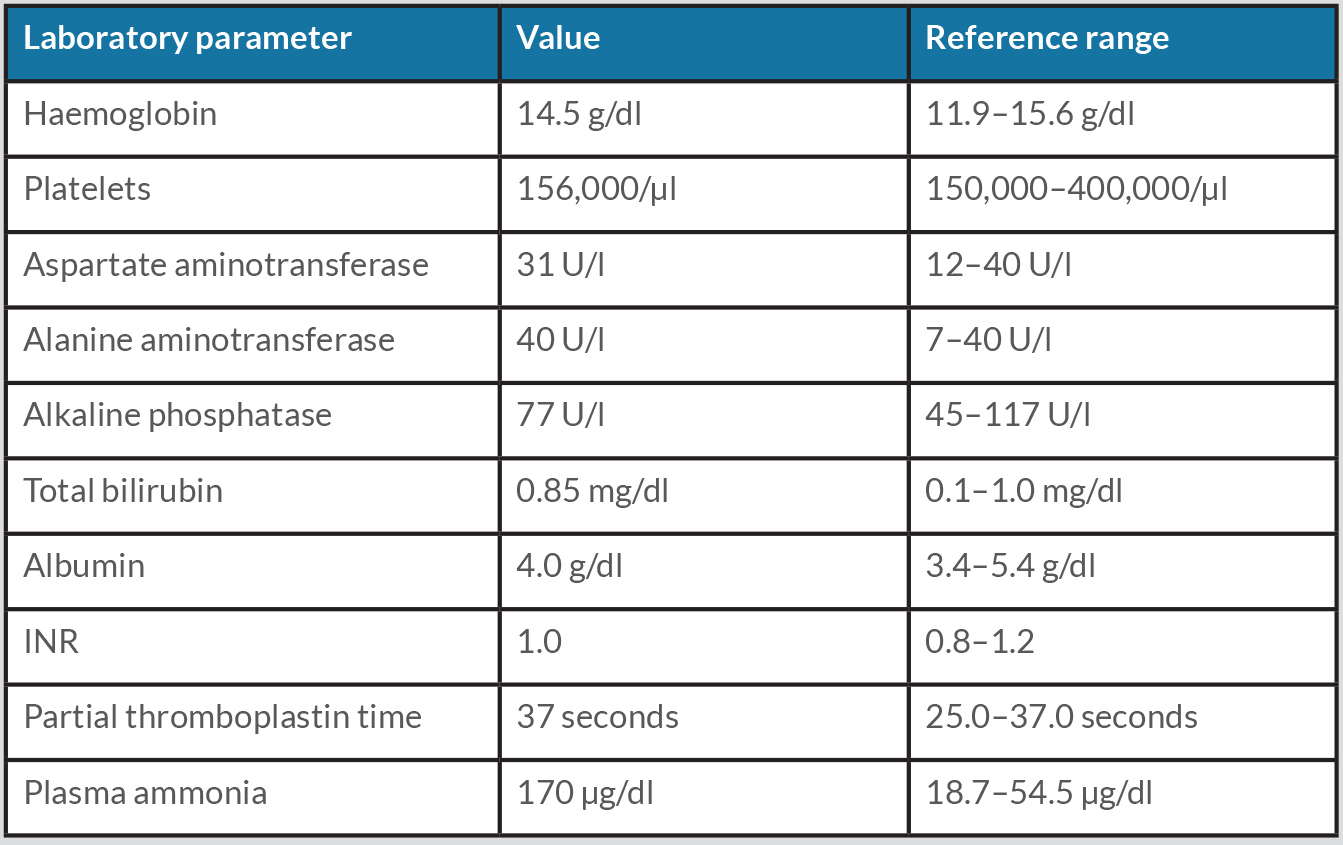
Table 1 (click to enlarge)
Figure 1. Axial (A) and coronal (B) CT images, in portal venous phase, showing a latero-lateral portocaval shunt of the left portal branch (yellow arrows)
Table 1. Laboratory results
DISCUSSION
The Abernethy malformation (AM) is a rare atypical connection between the portal vascular system and the systemic circulation, also known as congenital extrahepatic portosystemic shunt (CEPS), which was described for the first time by John Abernethy in 1793 [1].
According to the type of anastomosis and the status of the portal vein, it is classified into two types. Type I (end-to-side shunt) is characterized by complete extrahepatic shunting of portal blood into a systemic vein, with absent intrahepatic portal venous branches. Type II (side-to-side) are partial shunts between the portal vein and the systemic venous circulation, usually with the intrahepatic portal vein intact but hypoplastic [1, 2].
Its actual prevalence is unknown. However, as imaging diagnosis techniques advance, more cases have been reported. It is usually diagnosed in childhood, with 70% of the cases diagnosed before 18 years of age [3]. Nevertheless, mainly in type II shunts, symptoms can present later in life. It does not appear to have a sex predominance [1].
Abernethy malformations can be asymptomatic or associated with a broad spectrum of clinical manifestations, mainly associated with the portosystemic shunt [1].
One of the first manifestations can be hepatic encephalopathy of unknown cause [2]. This was the case with our patient, where Abernethy malformation type II was detected during the investigation. Due to portosystemic shunt, ammonia absorbed in the intestine bypasses hepatic metabolism and proceeds to the systemic circulation, reaching the brain.
Even though a direct connection between the portal and systemic venous systems exists, only 26% of patients with Abernethy malformation have hyperammonaemia, and just 15% of all patients present with hepatic encephalopathy [3]. In younger patients, hyperammonaemia can be present without encephalopathy. However, with age progression, clinical encephalopathy becomes more common. This may be explained by the diminished tolerance of the aging brain to high ammonia levels [3, 4].
Furthermore, hepatic encephalopathy also seems to depend on the shunt ratio. Uchino et al. showed that a shunt ratio above 60% (determined by portal scintigraphy) increased the risk of hepatic encephalopathy [4].
Both contrast CT and magnetic resonance angiography can be used to demonstrate extrahepatic portosystemic communication. They can accurately detect shunt anatomy and can characterize coexisting hepatic and extrahepatic lesions. When the results of these techniques are inconclusive, angiography can be utilized [1].
The therapeutic experience with Abernethy malformation is still limited. The treatment strategy is decided on a case-by-case basis and depends on shunt type, symptoms, complications and comorbidities. All symptomatic patients must be treated [1].
Generally, type I AM patients need liver transplantation, and type II AM patients are candidates for shunt closure, which can be performed either surgically or percutaneously. Before shunt closure, however, it is necessary to ensure adequate portal vein capacity and normal portal pressure to avoid post-procedure portal hypertension. In that regard, the intrahepatic portal system should be evaluated by angiography, using the shunt-occlusion test [1, 3].
The optimal timing for treatment is not defined. However, in most of the published literature, which is mainly focused on paediatric populations, early shunt closure is advised to avoid complications. In adulthood, there are fewer reported cases [1, 2].
The optimal therapeutic strategy is unclear in elderly patients with chronic hyperammonaemia and encephalopathy. Medical management of hepatic encephalopathy in these patients is identical to treating hepatic encephalopathy in cirrhotic patients and includes lactulose and non-absorbable antibiotics. In patients with encephalopathy controlled with treatment, the need for early closure of the shunt is not defined [2]. Recent studies report hepatic encephalopathy recurrence after shunt closure [3]. However, shunt closure seems to be more effective when no significant complications are established [1, 5].
Evidently, further in-depth studies are needed in order to establish the long-term prognosis of each therapeutic method.
In the reported case, the medical treatment was insufficient to control the hepatic encephalopathy completely, and so the patient was referred for shunt closure.
Despite being a rare cause of hepatic encephalopathy, Abernethy malformation should be considered in patients with unexplained hyperammonaemia. In the reported case, the patient had had symptoms for 2 years and was seen twice in the emergency room without the condition being recognized. Therefore, it is essential to raise awareness of Abernethy malformation since it is potentially reversible. In addition, early diagnosis and treatment may improve outcome [5].