ABSTRACT
Stevens-Johnson syndrome (SJS) is a severe dermatological disease classically characterized by erythematous target lesions and mucosal involvement. Fuchs syndrome is an incomplete presentation of SJS which has oral, conjunctival and genital manifestations but no skin lesions. To the best of our knowledge, our case of Fuchs syndrome in an 80-year-old man is the first such case related to herpes simplex virus (HSV)-1 infection to be described. Our patient quickly recovered following IVIG therapy, although specific treatment is still a topic of discussion. Research is required on this poorly understood dermatological disease to determine optimum therapy.
LEARNING POINTS
- We report a case of Fuchs syndrome in an elderly man after HSV-1 cheilitis.
- Therapy always includes discontinuation of the causative drug.
- Specific therapy for Stevens-Johnson syndrome and Fuchs syndrome is still a topic of discussion, although we noted marked improvement following the administration of IVIG therapy.
KEYWORDS
Fuchs syndrome, herpes simplex cheilitis, idiopathic thrombocytopenic purpura
INTRODUCTION
Stevens-Johnson syndrome (SJS) is a rare and potentially life-threatening immune-mediated mucocutaneous reaction[1–3]. Medications and infections such as herpes simplex virus (HSV) are the most common causes of the syndrome[1,4]. Although the combination of skin manifestations and mucosal involvement is the hallmark of SJS[5], patients can present with atypical manifestations showing strong mucosal involvement but with total or partial absence of skin lesions[6], making the diagnosis a clinical challenge. During the last two decades, several reports have described incomplete SJS (Fuchs syndrome) which did not include skin lesions but did have oral, conjunctival and genital manifestations.
We report a rare and severe case of Fuchs syndrome secondary to HSV-1 infection.
CASE DESCRIPTION
An 80-year-old man was transferred to our emergency department because of severe thrombocytopenia, petechiae as well as haematomas of the lower extremities, and macroscopic haematuria. The patient’s past medical history included idiopathic thrombocytopenic purpura (ITP), repeated deep venous thrombosis, and prostatic cancer in complete remission. Apart from edoxaban and eltrombopag, the patient had not received any other medications recently. Laboratory tests on admission showed a normal white cell count, anaemia and severe thrombocytopenia (1x109/l). C-reactive protein was normal.
We stopped anticoagulation therapy and started intravenous immunoglobulin (IVIG) 0.5 g/kg body weight. However, on hospital day 2, the patient developed chills and fever, with clinical and laboratory tests unable to identify the source of infection. The patient was started on prednisone 40 mg daily and empirical antibiotic treatment with ceftriaxone. IVIG treatment was stopped after 3 days of administration. The patient’s platelet count was normal at the end of treatment, but had decreased again by hospital day 18, so a new 3-day course of IVIG was initiated.
On hospital day 14, the patient had developed vesicles and bullae on an erythematous background suggestive of herpes simplex cheilitis covering the entire lower lip and the left part of the upper lip. PCR confirmed the diagnosis of HSV-1 cheilitis and the patient was started on famciclovir 500 mg three times per day. Despite antiviral treatment and contrary to our expectations, the clinical situation worsened with the development of extensive erythema with erosions and ulcers on labial and oral mucosa (Fig. 1). However, ocular or genital involvement and cutaneous manifestations were not observed.
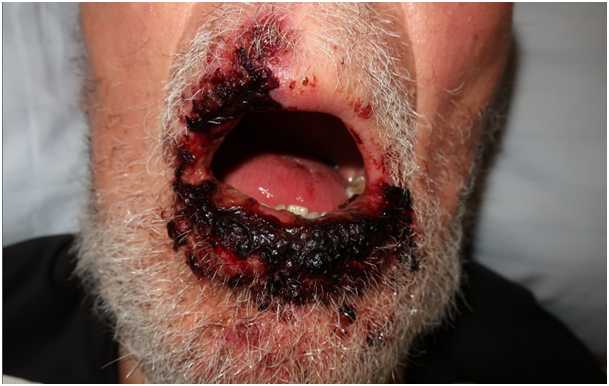
Figure 1a (click to enlarge)
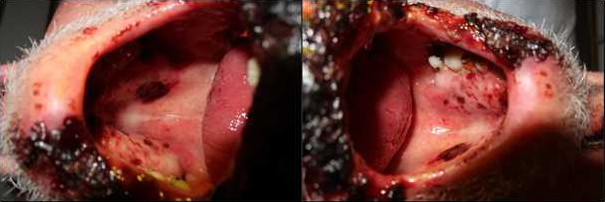
Figure 1b (click to enlarge)
Figure 1. Acute presentation of a patient with Fuchs syndrome with extensive involvement of the oral mucosa secondary to severe HSV-1 cheilitis
Fuchs syndrome was diagnosed based on the clinical appearance in combination with the associated HSV-1 infection. Accordingly, systemic treatment with prednisolone and antiviral therapy was continued and local symptomatic therapy with acetylsalicylate and chlorhexidine was initiated. A remarkable improvement was observed after IVIG was reinstated on hospital day 18, with almost complete resolution of the lesions (Fig. 2). After treatment, the patient did not experience any new outbreaks and was discharged after 25 days of hospitalization.
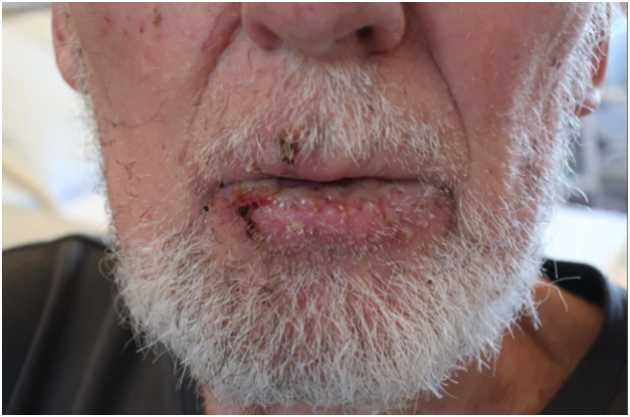
Figure 2 (click to enlarge)
Figure 2. Remarkable improvement with almost complete resolution of the lesions
DISCUSSION
SJS is a rare and severe immune-mediated mucocutaneous reaction[1,2]. The syndrome was first described in 1922 by Albert Mason Stevens and Frank Chambliss Johnson in a report of two paediatric patients with fever, stomatitis and ocular mucosal lesions[3]. Medications such as antimicrobials, antiepileptics, allopurinol and non-steroidal anti-inflammatory drugs are the main triggers of SJS[4]. Other important causes, especially in the paediatric population, include infectious agents such as Mycoplasma pneumonia and herpes simplex virus[1].
The cutaneous symptoms of SJS are frequently described as erythematous or violaceous patches, atypical targeoid lesions, erosions, bullae and ulcerative manifestations after a prodromal phase of fever and flu-like clinical signs and symptoms which last 1–3 days[4,5]. Almost 80% of patients with SJS develop mucosal involvement, with oral mucosa being more commonly involved than the eyes and genitalia[4,5]. Atypical presentations are primarily described in the paediatric population[6] and are frequently associated with M. pneumonia infection[7]. Although herpes simplex is a typical SJS trigger, to the best of our knowledge, the presented case of Fuchs syndrome is the first to be associated with HSV. The diagnosis of HSV-1 cheilitis was confirmed by PCR and, contrary to our expectations, the clinical findings worsened dramatically despite antiviral therapy with famciclovir. The characteristic erythematous, erosive and ulcerative lesions of the entire oral cavity suggested the diagnosis of atypical SJS. Further, the differential diagnosis of extensive HSV-1 infection was improbable due to large extension in an immunocompetent patient with reactivation (non-primary infection) of HSV-1 and the initial improvement under antiviral therapy.
There is increasing evidence that a specific human leukocyte antigen (HLA) type is associated with the development of SJS[1,5]. The distinctive findings of full-thickness epidermal necrosis is considered to be of apoptotic origin[1,4,5]. Apoptosis is induced directly and indirectly by specific cytotoxic CD8 Τ-cell populations and natural killer cells via release of soluble mediators[4].
Given the high morbidity and mortality rates of SJS and the potential for rapid progression[4], immediate discontinuation of suspected causative drugs and initiation of supportive care including fluid and nutrition support, wound care, pain relief, and control of renal function, electrolytes and the airway, are crucial in the initial phase of SJS[1,4,5]. Adjunctive therapies such as systemic corticosteroids, IVIG and tumour necrosis factor (TNF) inhibitors have been proposed as SJS therapy[4,8]. The role of systemic corticosteroids remains controversial as there is evidence that they may increase morbidity and mortality, probably due to increased risk of infection[2,9]. Although IVIG has been widely administered to patients with SJS[1,4,5], the data supporting its use are contradictory[5,10]. However, in our patient, initiation of IVIG due to relapse of severe thrombocytopenia resulted in a rapid improvement in clinical findings.
In conclusion, this case report describes a patient with SJS due to herpes simplex infection, who presented with severe involvement of the oral mucosa but no cutaneous lesions (Fuchs syndrome). We demonstrate that Fuchs syndrome can present with isolated oral mucosal lesions but without skin, ocular or genital manifestations, and found that clinical improvement occurred following the initiation of IVIG therapy.