ABSTRACT
Introduction: Strokes are common but can be caused by a rare illness. Moyamoya disease (MMD) justifies a family assessment because of its hereditary nature and the availability of new therapies.
Case description: A 42-year-old man was admitted because of convulsions with sensorimotor deficit due to a massive cerebral haemorrhage caused by MMD. The fact that the patient died suggested his children should be screened.
Discussion: MMD is rare and its consequences disastrous. Many cases in both children and adults have been described. Investigations should be carried out when the diagnosis is suspected, and, if confirmed, the family should be screened given the genetic nature of some forms of the disease. Effective and increasingly personalized therapeutic solutions are available.
Conclusions: A minority of strokes are caused by rare diseases including MMD. Our current knowledge of this pathology and the treatments available justify a family assessment when the clinical or family context requires it.
LEARNING POINTS
- Strokes are frequent but can reveal rare conditions, including Moyamoya disease
- The families of patients should be screened in order to avoid morbidity and mortality.
- The diagnosis is not necessarily fatal as there are various promising therapeutic solutions, which should encourage further research.
KEYWORDS
Stroke, Moyamoya disease, neurology, genetics
INTRODUCTION
Stroke is a major public health problem with 80% of ischaemic and 20% of haemorrhagic strokes caused by high blood pressure as the main risk factor [1, 2]. The many aetiologies include Moyamoya disease (MMD), a rare entity characterized by chronic stenosis of the internal carotid artery and its branches [3]. We report the case of a 42-year-old patient whose diagnosis was revealed by massive cerebral bleeding.
CASE DESCRIPTION
Our Emergency Department (ED) team received a call from a sports hall regarding an inaugural seizure in a 42-year-old Caucasian man. Upon our arrival, the patient was unconscious. Witnesses reported he had had a severe headache with deformation of the face, followed by loss of consciousness with generalized convulsions. This had happened 30 minutes previously, while the patient was playing badminton. The convulsions stopped after a few minutes.
The patient had high blood pressure and hypercholesterolemia, neither of which were treated. A history of stroke was mentioned by his mother. The patient had no allergies or addictions. He was married and the father of two children.
On site, the patient was unconscious with a Glasgow Coma Scale (GCS) score of 3. There was urinary incontinence, tongue biting and anisocoria (left mydriasis). Lower right facial paralysis was obvious, but no other sensory-motor anomalies were seen in the postictal phase. Examination revealed a symmetrical blood pressure (BP) at 205/100 mmHg, with a heart rate (HR) of 95 bpm, a respiratory rate (RR) of 24 per minute and oxygen saturation of 99%. The patient’s glycaemic index was 1.25 g/l and his temperature was 35.9°C.
The clinical picture prompted intubation with succinylcholine and etomidate, followed by sedation with midazolam and sufentanil. In light of the anisocoria indicating intracranial hypertension, 4 g of hypertonic salt serum (10%) was administered. During the journey to hospital, factors indicating secondary brain injury were monitored: normocapnia and normoxia were maintained and permissive hypertension remained below 220/120 mmHg. No hypotensive therapy was necessary.
In the ED, transcranial Doppler ultrasound (TCD) confirmed the absence of cerebral flow on the left side (diastolic perfusion rate of zero), with no window on the right. Cerebral magnetic resonance imaging (MRI), 2 hours after the first symptoms had developed, confirmed the presence of a large haematoma in the left hemisphere, complicated by subfalcine and temporal engagements. Many vascular abnormalities confirmed the presence of MMD (Fig. 1).
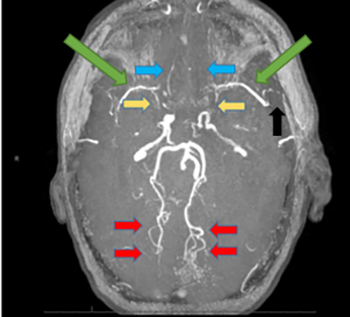
Figure 1 (click to enlarge)
Figure 1. Cerebral angio-MRI, in time-of-flight (TOF) sequence, showing small Sylvian vessels bilaterally (green arrows) as well as an absence of opacification of the M1 portions of both Sylvian arteries (yellow arrows). Several branches of the left Sylvian artery are occluded (black arrow). The left anterior cerebral artery is not opacified (blue arrows). In compensation, the posterior cerebral circulation is highly developed (red arrows)
The patient’s neurological state rapidly deteriorated (abolition of brainstem reflexes after MRI) and collegial discussion ultimately led to a decision to abstain from surgery with therapeutic regression. As no contraindications to organ donation were identified, the procedure was authorized. Family screening and support for family members were also offered.
DISCUSSION
TMMD is a cerebral vasculopathy, usually bilateral, affecting the internal carotids and their branches, particularly the circle of Willis. Chronic stenosing and occlusive problems stimulate the development of capillary replacements at the base of the brain. This modifies the angiography results, which are characterized by a ‘puffs of smoke’ appearance, from which the name Moyamoya derives [3]. Initially described in 1957 in Japan, the various forms were classified in 1969. The disease is more widespread in Asian countries (0.3 to 0.9/100,000 Japanese cases in 2014) but is also described in Europe and America, with a prevalence 10–20 times lower [4].
The most commonly reported symptomatology is transient ischaemic attack (TIA) in childhood. Cerebral haemorrhage as the inaugural manifestation is significantly higher in adults (and often fatal, as in this patient) [3, 5]. The literature describes two peaks of incidence, at around 5 and 40 years of age. Haemorrhagic manifestations seem to be due to the rupture of collateral pathological vessels (weakened and/or saccular aneurysm) [3, 4]. There are several other clinical pictures including syncope, convulsions, headaches, choreiform movements and cognitive disorders [4, 5].
Diagnosis is based on imaging, which usually combines angiography (anatomy) and nuclear medicine (haemodynamic impact). However, there are many less invasive and promising techniques including MRI, which has become fundamental to diagnosis and post-operative follow-up, particularly in paediatric patients. Its different modes of use make it a technique of choice for anatomical and haemodynamic investigation and also for the study of cerebral vascular reserve [3, 4]. Computed tomography (CT) is used for diagnosis and post-operative follow-up but raises the problem of irradiation and contrast toxicity for repetitive follow-up. TCD ultrasound at the patient's bedside allows haemodynamic study by cerebral perfusion rates in different arteries. In our patient, this examination immediately suggested a very poor prognosis. It is important for monitoring the immediate post-operative evolution after revascularization. Nevertheless, it remains operator-dependent and does not cover the entire vascular system, and therefore remains complementary to other imaging [4].
The hypothesis of a genetic origin is widely accepted, and the involvement of the RNF213 gene on chromosome 17 [6, 7]. One Asian-specific mutation p.R4810K is frequent in East Asian MMD patients and increases susceptibility to MMD with a dose-dependent effect on clinical phenotype severity. This variant, observed in 1–2% of the general East Asian population, may explain the higher incidence of MMD in Asia but its penetrance is probably low as there are many unaffected carriers. Other variants of RNF213 and rare variants in other genes have been found in MMD patients worldwide [7]. Some 10–15% of patients have affected family members [3], and autosomal dominant modes of transmission with incomplete penetrance have been identified in some pedigrees [5]. Recently, advances in genetics have allowed a major breakthrough, but knowledge remains limited and we need other studies in the future. Furthermore, the family history of our patient prompted genetic assessment of his children. To date, not all pathological variants have been identified, the modes of transmission are unknown, and penetrance and symptomatology are variable. In addition, environmental factors are most likely involved, which complicates investigation.
Finally, abnormalities in the electroencephalograms of symptomatic patients have been identified, particularly slow waves of large amplitude in the posterior (slow activity P) and centro-temporal (slow activity CT) zones [3]. This examination can suggest the disease but does not constitute a diagnosis.
A check-up in familial or symptomatic cases is indicated since there are many therapeutic possibilities, in particular vascular bypass surgery by direct (temporal artery anastomosis) or indirect (non-anastomotic from distant cerebral branches) methods [3]. Direct bridging allows instantaneous improvement of blood flow and is preferred in adults, who have a larger calibre Sylvian artery which is therefore more accessible. The indirect technique (several combinations) is used more in paediatric patients and stimulates the formation of new collaterals after a few months [3, 8]. The results are excellent for reducing haemorrhagic stroke. The benefit is less obvious in ischaemic stroke, but other studies should confirm it [8]. In addition, augmented reality techniques (real-time image superposition) are encouraging for personalized surgical treatment for each patient (adapted craniotomies, directed vascular dissections, better identification of anastomotic vessels). In this minimally invasive surgery, the contribution of endovascular techniques is much less promising [9]. Indeed in the ischaemic context, the placement of an endovascular stent or the dilation of fragile arteries is dangerous. On the other hand, the small size of the arteries and the length of the stenoses are often contraindications, especially in paediatric patients. Finally, the long-term efficacy in a progressive disease is unknown. In the haemorrhagic context, embolization of an aneurysm involves the risk of occlusion of the distal artery. At this time, this surgery is not recommended [3].
Finally, conservative techniques including anti-platelets for ischaemic forms are recommended. The use of anticonvulsive therapy (epilepsy) or calcium antagonists (migraine) depends on the clinic [10].
CONCLUSIONS
This case involved a Caucasian patient who died from massive cerebral haemorrhage due to MMD. This rare disease is increasingly well described but nevertheless remains little known. Neurological symptoms in childhood and/or stroke at a young age without identified risk factors should motivate investigation. Family screening including imaging and genetic assessment should be considered if imaging is suggestive. Current medicine offers various therapeutic possibilities justifying a diagnostic work-up when the context (clinical and/or family) suggests it. If a clear prognosis could be made on a genetic basis, this could allow adapted therapeutic intervention for all familial cases.