ABSTRACT
Polymyositis (PM) is usually associated to other autoimmune or connective tissue diseases. The authors report the case of a 59-year-old man with pulmonary fibrosis, who presented with constitutional symptoms and gradually developed proximal muscle weakness, Raynaud phenomenon, and dysphagia. Besides creatine kinase (CK) elevation, he had positive anti-Polymyositis-Scleromyositis (PM-Scl) and anti-Sjögren's-syndrome A (SSA) antibodies. Nailfold capillaroscopy showed a scleroderma pattern and muscle biopsy revealed necrosis, regeneration of muscle fibers, and inflammatory infiltrate. Prednisolone was started, with great improvement. Taking into account the overlap features between PM and systemic sclerosis sine scleroderma, it is important to closely monitor the patient for signs of pulmonary and cardiac decompensation.
LEARNING POINTS
- Polymyositis (PM) may be associated with connective tissue diseases such as systemic sclerosis, including its variant without skin involvement.
- Necrotizing muscle fibers are typically found in patients with overlap syndrome, in opposition to patients only with polymyositis.
- PM-Scl antibodies are associated to a good response to corticoids.
KEYWORDS
Polymyositis, systemic sclerosis sine scleroderma, overlap syndrome.
INTRODUCTION
In the complex field of connective tissue diseases and autoimmunity it is known that many patients can not be categorized into one clinical entity. Polymyositis (PM) is an inflammatory myopathy that may have a heterogeneous clinical presentation but usually courses with subacute proximal muscle weakness and creatine kinase (CK) elevation. It is rare as a single entity. Actually, it is often seen in association with systemic autoimmune disorders or connective tissue diseases. Systemic sclerosis-polymyositis (SSc-PM) overlap syndromes are usually anti-PM-Scleromyositis (Scl) antibody positive and these are a marker of good prognosis. 1.
CASE REPORT
A 59-year-old man was referred to Internal Medicine for consultation due to asthenia, anorexia and weight loss (12% of body weight) lasting for five months. He also had rectal bleeding. Two months prior, an exudative pleural effusion (4100 cells, 78% neutrophils) was diagnosed after an episode of dyspnoea, fever and pleuritic chest pain. Besides pleural effusion, thorax computed tomography (CT) scan showed a small emphysematous bulla in the left upper-lobe and lower-lobe bilateral pulmonary fibrosis. Echocardiography and bronchofibroscopy were normal and pulmonary function tests showed small airways obstruction. By the time he presented to our consultation, his blood tests showed elevated liver enzymes (AST 121 U/L, ALT 187 U/L, GGT 130 U/L) and normocytic normochromic anaemia (Hb 10,6 g/dl, VGM 90,7 fl). His abdominal ultrasound was normal, viral serologies were negative, and colonoscopy showed 4 polyps, one of them a tubullovilous adenoma.
Three months later, he started complaining of myalgia of the scapular and pelvic girdles and described Raynaud phenomenon, without skin lesions. A 5-fold CK elevation was detected, with normal sedimentation rate. The patient was tested for autoantibodies and was anti-PM-Scl 100/75, anti-SSA and anti-nucleolar positive. Antibodies to aminoacyl-transfer ribonucleic acid (RNA) synthetases was not detected. A nailfold capillaroscopy was preformed and showed moderate loss and dilated capillaries, frequent haemorrhages, and mild disorganisation of the capillary architecture as well as neovascularization, suggesting an autoimmune disease with scleroderma pattern or neoplasm (Fig. 1.)
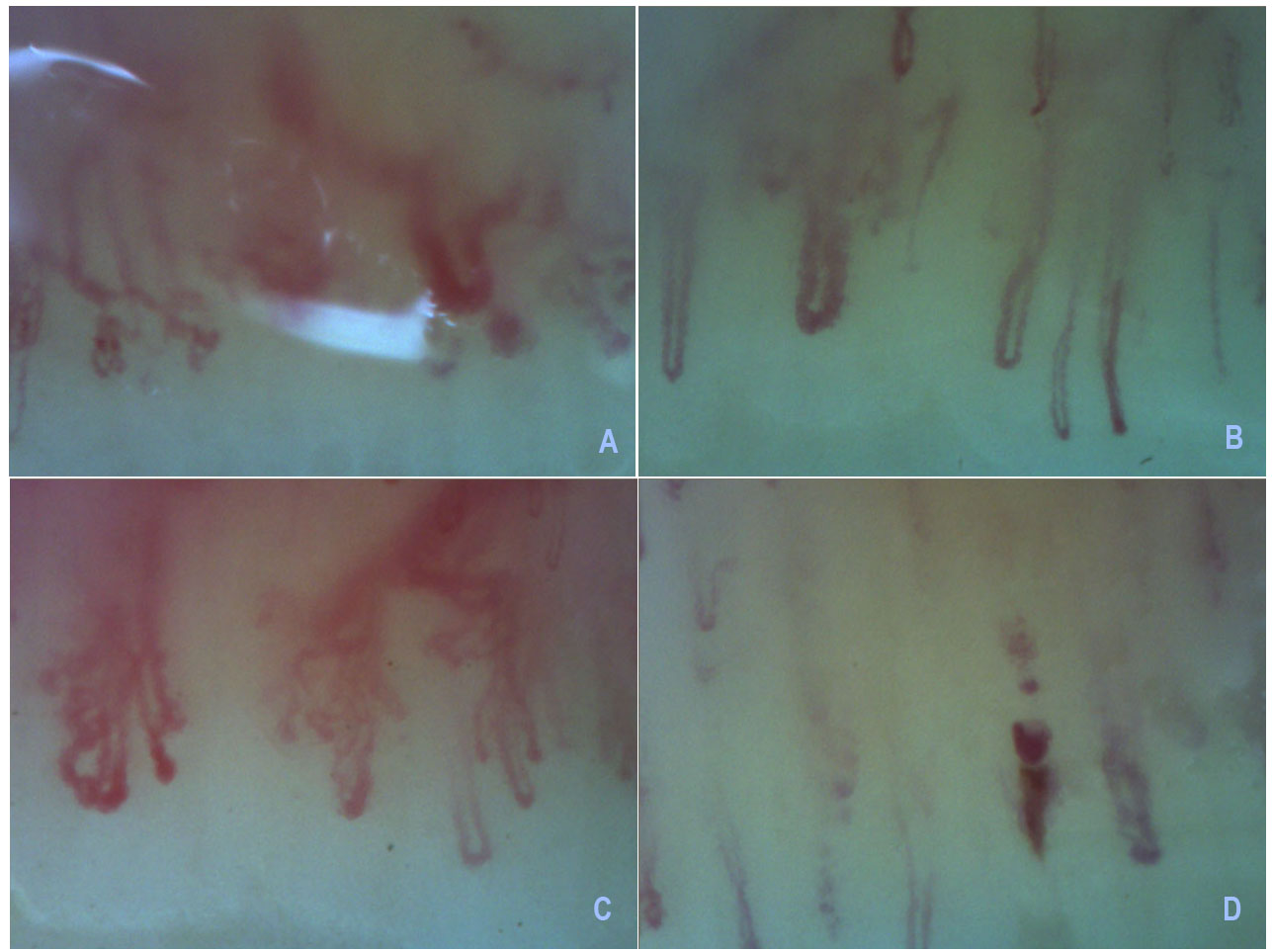
Figure 1 (click to enlarge)
Figure 1. A, B, C - Severe loss of capillaries with avascular areas, interstitial edema, enlarged loops, disorganisation of the normal capillary architecture and ramified capillaries;
D – hemorrhage.
There was no suspicious uptake of fluorodeoxyglucose (18 F-FDG) on positron emission tomography scan, and electromyography was normal. The patient then started complaining of dysphagia, leading to a larger weight loss and myalgia, which became unbearable, restricting his ability to perform daily living tasks. He was admitted to Internal Medicine ward and esophageal manometry was requested, showing normal relaxation of the gastroesophageal junction, weak peristalsis, and large peristaltic defects. The barium esophagogram disclosed deficient propulsion of tongue contrast accumulation in valleculae and episodes of laryngeal penetration. The muscle biopsy revealed necrosis and regeneration of muscle fibers and inflammatory infiltrate composed of B and T lymphocytes, plasmocytes and histiocytic cells, a pattern compatible with polymyositis. The patient was started on prednisolone 40 mg id with progressive clinical improvement. Corticoid was tapered and two years after the diagnosis he was asymptomatic with a treatment of prednisolone 2,5mg id, leading to a recovery of functionality and weight. Pulmonary function stabilized and the patient is being closely monitored.
DISCUSSION
The authors present this case highlighting the importance of recognizing the possibility of overlapping features between polymyositis and systemic sclerosis, and excluding concomitant malignancy.
Although the incidence of malignancy is only slightly increased in polymyositis, constitutional symptoms and rectal bleeding presented initially, along with anaemia and the evidence of neovascularization in nailfold capillaroscopy, justified the search for occult malignant disease1. Along with weakness in proximal muscles, polymyositis can present with dysphagia, mostly pharyngeal, in 10-15% of patients and with pulmonary fibrosis in 5 to 10%1. Even though changes are usually observed in electromyography (EMG), nearly 10% of patients have a normal EMG, possibly due to insufficient sampling1.
Our patient had positive anti-SSA antibody, usually associated to myositis, but also anti-PM-Scl antibodies, characteristic of SSc-PM overlap2.
Nailfold capillaroscopy, as a non-invasive method to assess skin microvasculature, is used to discriminate primary from secondary Raynaud phenomenon and is particularly important in the evaluation of patients with systemic sclerosis spectrum diseases3. Raynaud’s phenomenon is almost universal in patients with scleroderma and may be present in 20-40% of patients with myositis2. The SD pattern was found to be present in 20–60% of patients with DM or PM and to correlate with interstitial pulmonary involvement1.
A recent study comparing myopathologic aspects of patients with scleroderma-PM overlap compared with a population of patients with systemic sclerosis and PM revealed that necrotizing muscle fibres are found in almost all patients with the overlap syndrome, in opposition to patients with PM4. The finding of this kind of fibres in our patient, favours the diagnosis of an overlap of PM with a variant form of limited systemic sclerosis without scleroderma–systemic sclerosis sine scleroderma.
Although our patient did not have apparent cardiac involvement, cardiac complications are known to be a major cause of death in patients with SSc-PM overlap4.
In what concerns to treatment and outcome, our patient had a good response to prednisolone which is consistent with literature, as response to small doses of corticosteroids has described in patients with PM-Scl overlap2.
Although this patient presented mainly with clinical features of PM and had a good response to corticoid therapy, the presence of pulmonary fibrosis, the auto-immunity and nailfold capilaroscopy pattern as well as myopathologic findings support the diagnosis of an overlap syndrome. This hypothesis reinforces the need for close monitoring of cardiac and pulmonary complications in the future.