ABSTRACT
It is rare for IgM multiple myeloma (MM) and mantle cell lymphoma (MCL) to coexist. Furthermore, it is challenging to demonstrate if there are two distinct types of neoplasia or if plasma cell differentiation of MCL is present. We discuss the case of a patient concomitantly diagnosed with MCL and IgM MM, and the subsequent diagnostic and management difficulties, and the positive treatment outcome.
LEARNING POINTS
- Due to the rarity of simultaneous multiple myeloma (MM) and mantle cell lymphoma (MCL), it is challenging to investigate a possible association.
- This report will draw attention to the condition's rarity, diagnostic and treatment hurdles, and hopefully inspire future research into therapeutic alternatives.
- Several recent developments indicate a bright future for treating refractory malignancies such as MM and MCL, such as the advent of chimeric antigen receptor T-cell (CAR T-cell) therapy.
KEYWORDS
IgM multiple myeloma, mantle cell lymphoma, CAR T-cell therapy
INTRODUCTION
Multiple myeloma (MM) and mantle cell lymphoma (MCL) are well-known B-cell malignancies with an extensive global disease burden [1]. Only a few cases of IgM MM have been described in the literature, and MM occurring together with MCL is even rarer. In addition, plasma cells are one of the neoplastic components of several types of non-Hodgkin’s lymphoma (NHL), although they have only been rarely seen in MCL cases [2]. Naturally, this rare pathological duality presents significant treatment challenges. We present the case of a 55-year-old man with a very rare concomitant diagnosis of IgM MM and MCL, and discuss its management.
CASE DESCRIPTION
The patient was a 55-year-old man with no significant past medical history who was seen by his primary care physician after developing an episode of diverticulitis. At that time the patient was noted to have low or low-normal values on a complete blood count (CBC), including white blood cells (WBC) 3.2 (103/µl) with an absolute neutrophil count (ANC) of 1500 (103/µl), haemoglobin (Hb) 12.4 g/dl with mean corpuscular volume (MCV) 94 fl, and platelets (PLT) 184,000 (103/µl). Approximately 2 months after that, the patient was given an ‘aggressive hug’ by his son and immediately developed rib pain. The patient had another occurrence of hugging-induced rib pain 3 months later at which time a routine CBC showed worsening pancytopenia and a chemistry panel demonstrated an elevated total protein level of 10.1 g/dl. CBC showed WBC 3.6 (103/µl), ANC 1400, Hb 8.0 g/dl, MCV 98 fl, and PLT 98,000 (103/µl). Serum protein electrophoresis demonstrated an M-spike of 4.6 g/dl in the gamma region with immunofixation confirming the presence of an IgM monoclonal protein with kappa light chain specificity, with IgM level elevated to 5791 mg/dl. Serum beta-2 microglobulin was elevated at 2.9 mg/l. Viscosity was also elevated at 5.2. Given these findings, the patient was referred to the haematology clinic.
Upon presentation, the patient endorsed unintentional weight loss of around 10 pounds, joint pains, night sweats, and generalized fatigue over 2–3 months. He denied a history of smoking and drank alcohol socially. His family history was significant for lung cancer in his mother, who had died at age 75, and unspecified neck/throat cancer in his father, who had died at age 59. Vital signs were noted to be within normal limits. Physical examination was notable only for a palpable nodule on the patient’s right shoulder. Bone marrow (BM) aspirate and biopsy demonstrated CD5-positive mature B-cell lymphoma, consistent with MCL accounting for 15-20% of overall cellularity as well as an abnormal plasma cell (PC) population accounting for 80–85% of overall cellularity (Fig. 1).
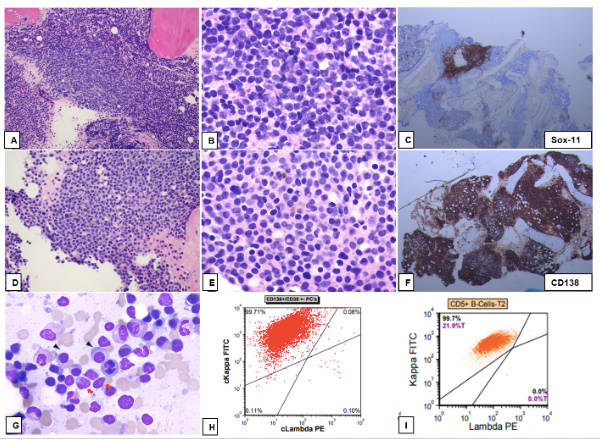
Figure 1 (click to enlarge)
Figure 1. Bone marrow specimen with concurrent myeloma and mantle cell lymphoma (MCL). (A–C) Area of marrow with nodular involvement by MCL (A; H&E, 100×); lymphoma cells show irregular nuclear contours and scant amount of cytoplasm (B; H&E, 400×) and are immunoreactive for SOX-11 (C; immunostain, 40×). (D–F) Plasma cell neoplasm with small plasma cells (D; H&E,100×). Some plasma cells display Dutcher bodies (E; H&E, 400×). Diffuse plasma infiltrate is highlighted by CD138 immunostain (F; 40×). (G) Bone marrow aspirate smear with many plasma cells with predominant small size (arrowhead) and fewer lymphoma cells with scant cytoplasm and irregular nuclear contours (red arrows) (G; Wright-Giemsa stain, 1000×). (H, I) Flow cytometry of bone marrow aspirate showing kappa light chain restricted plasma cell population gated by CD138+/CD38+ (H) and kappa restricted CD5+ B-cell population (I)
Background marrow showed decreased maturing trilineage haematopoiesis. Cyclin D1 was positive and monoclonal protein was concordant with the abnormal B cells and PC, creating uncertainty as to whether this represented separate or related processes. The MCL population was noted to be CD20+, CD79+, CD5+, cyclin D1+, SOX-11+ and MUM1–. The PC were CD138+, MUM1+, cyclin D1+, CD20 partial, PAX5–, SOX-11, CD79a–, CD56–, CD117–, IgM weakly positive, and IgG negative. Fluorescent in situ hybridization (FISH) was notable for 13q- and t[11;14]. IGHV was also noted to be mutated while MYD88 mutation analysis was negative. The patient had a PET scan in June 2021 which showed multiple subcutaneous lesions, lymphadenopathy (LAD) in the left axillary region with SUV 1.6, retroperitoneal, mesenteric, and pelvic sidewall LAD up to 1.9 cm with SUV of 2.8, spleen uptake of 2.0 compared to liver of 1.8, and homogeneous BM activity with SUV of 3.3. The patient then underwent an excisional biopsy of a right shoulder subcutaneous lesion. This biopsy showed lymphoma cells positive for CD20, cyclin D1 and SOX-11 (Fig. 2).
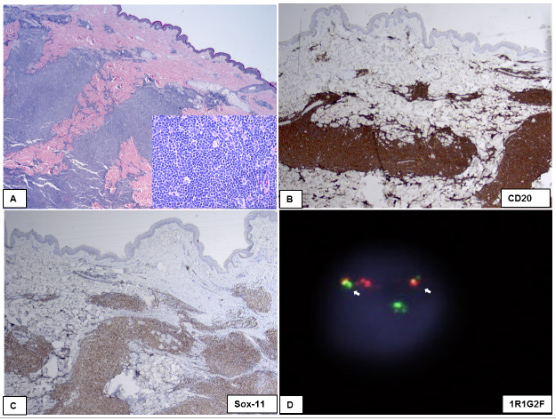
Figure 2 (click to enlarge)
Figure 2. Mantle cell lymphoma involving dermis and subcutaneous tissue (A; H&E stain, 40×; inset 200×). The lymphoma cells show cytoplasmic immunoreactivity for CD20 (B; 40×) and nuclear immunoreactivity for SOX-11 (C; 40×). (D) FISH study using a dual colour, dual fusion CCND1(BCL1)/IGH probe set showed an abnormal dual fusion signal pattern of 1R1G2F, indicative of CCND1(BCL-1)/IGH fusion (white arrows show the fusion signals).
The proliferation rate by Ki-67 was about 40% and the cells exhibited fine chromatin, frequent apoptosis, and increased mitotic activity, supporting a blastoid variant of MCL. The lymphoma involved dermis and subcutaneous tissue and extended to the resection margins. Flow cytometry immunophenotyping on a portion of the tissue showed a CD5 positive kappa light chain restricted B-cell population suggestive of MCL. The diagnostic challenge here was due to the cyclin D1 positivity and the same type of monoclonal protein expression by both abnormal B-cells and PC, making it difficult to ascertain whether abnormal PC represented a separate PC neoplasm unrelated to B-cell lymphoma or if this process represented PC differentiation of MCL. In the end, a separate PC neoplasm unrelated to MCL was favoured due to the aberrant phenotype of PC (CD45– and CD19–). Next generation sequencing (NGS) from blood came back positive for p53 and two ATM mutations, with some uncertainty as to whether the p53 mutation was related to MCL, MM or both. The case was discussed at multidisciplinary tumour board and given the extent of the BM involvement by a plasma cell dyscrasia with progressive cytopenias with increasing viscosity, treatment with KCD (carfilzomib, cyclophosphamide, Decadron) with rituximab and lenalidomide was recommended. The patient completed four cycles of R-KCD + lenalidomide in total and was then admitted for inpatient high-dose chemotherapy with carmustine, etoposide, cytarabine, melphalan (BEAM) and autologous stem cell rescue (ASCR). The patient tolerated the procedure well, eventually having neutrophil engraftment in early March before being discharged home.
DISCUSSION
It is exceptionally rare for MCL and plasma cell dyscrasia to coexist. Even though both cell populations express cyclin D, the abnormal phenotype of CD19 negative and CD45 negative PC suggested a distinct synchronous neoplasm independent from MCL with clonal PC differentiation. PC can be seen as a neoplastic component of several types of NHL, although they have only been rarely seen in MCL cases [2]. The diagnostic challenges in clearly delineating between these two pathologies unfortunately also necessitates a more complex treatment plan as well, for which specific trial data is relatively scarce.
MCL occurs in naive B cells' primary lymphoid follicles or the mantle zone of secondary lymphoid follicles that have not gone through the germinal centre (GC) process [3]. In the GC, the cells undergo somatic hypermutation and class switch recombination to undergo further proliferation and differentiation [4]. The GC B cells then have a higher chance of surviving and differentiating into PC or memory B cells [5]. As a result, plasmacytic differentiation is not uncommon in lymphomas like follicular, marginal and diffuse large B-cell lymphomas, which emerge from B cells at either the GC or post-GC developmental stage [6–8]. On the other hand, plasmacytic differentiation is occasionally seen in pre-GC lymphomas like MCL. A study found that clonal PC differentiation can occur in some cases of MCL, suggesting that PC and MCL cells can originate from the same B-cell clone [2, 4]. It was suggested that clonal PCs could be found in the MCL tumour microenvironment and that their frequency in MCL may be higher than previously considered [4]. The presence of t[11;14][q13;q32] in both MCL cells and clonal PC was previously thought to indicate the presence of a common B-cell clone. However, the presence of the same translocation is insufficient to establish common clonality, as it is also seen in non-hyperdiploid plasma cell myeloma [4, 9]. Differentiating between MCL with a concomitant plasma cell neoplasm and MCL with plasmacytic differentiation is critical since treatment and prognosis are predicted to differ [4].
MCL is a generally aggressive cancer because it is prone to developing abnormalities in the cell cycle or cell survival pathways [10, 11]. It is also primarily associated with t[11;14][q13;q32] [11]. B cells in classic MCL do not penetrate the GC but express the transcription factor SOX11[10]. Leukaemic non-nodal MCL arises through the GC with IGHV mutation, has a more indolent course and genetic stability throughout time, and has limited or no SOX11 expression [10, 11]. With unmutated IGHV gene rearrangement and SOX11 overexpression, as well as a higher degree of genomic instability, nodal MCL has an aggressive course [11]. Blastoid cytology is linked to a shorter progression-free survival (PFS) and shorter overall survival (OS), with only the latter being independent of the MIPI (MCL International Prognostic Index) [10]. The patient was found to have positive SOX11 expression, high Ki-67% activity of about 40%, and pathology suggesting blastoid variant. TP53 mutations associated with blastoid morphology had inferior outcomes compared with TP-53 unmutated cases [10, 12]. They were associated with high Ki-67, high-risk MIPI, and MIPI-c [10]. The median time to therapy was 12 months, with individuals treated within 3 months of diagnosis having a higher survival rate [10]. Frontline treatment for MCL is determined by the patient's age and condition and may include aggressive chemotherapy such as rituximab and cytarabine and consolidation ASCT [10]. Our patient had an excellent response to the R-KCD regimen with lenalidomide.
Various advancements in newer techniques, such as the introduction of chimeric antigen receptor T-cell (CAR T-cell) therapy, show promise for the treatment of refractory cancers including MM and MCL [13]. It will be fascinating to see where the future leads us.