ABSTRACT
Cerebral amyloid angiopathy (CAA) is characterised by β-amyloid deposition in the walls of small to medium sized arteries of the cerebral cortex and the leptomeninges. In a significant proportion of patients, CAA is the probable cause of non-traumatic primary cerebral haemorrhage, particularly in those who are over 55 years of age and have controlled blood pressure. Cerebral amyloid angiopathy-related inflammation (CAA-ri) is an uncommon and aggressive subtype of CAA, which is thought to be caused by an immune reaction to the deposits of β-amyloid. It has a variety of presentations that can mimic other focal and diffuse neurological disorders. Radiographically, its classic presentation is asymmetric cortical or subcortical white matter hyperintense foci due to multiple microhaemorrhages on T2-weighted or fluid attenuated inversion recovery (FLAIR) images. Although definite diagnosis requires brain and leptomeningeal biopsy, diagnostic criteria for probable CAA-ri based on a combination of clinical and radiological features were validated in 2015. We describe a patient with probable CAA-ri mimicking stroke and review the clinical and radiological features important for a proper differential diagnosis between ischaemic stroke (IS) and CAA-ri, and its subsequent appropriate treatment.
LEARNING POINTS
- MRI is a crucial tool for the diagnostic evaluation of cerebral amyloid angiopathy-related inflammation (CAA-ri).
- A high index of suspicion and awareness of CAA-ri is necessary for correct diagnosis in stroke-like presentations of the condition.
- The treatment of choice for CAA-ri is empirical corticosteroid therapy, which is associated with clinical and radiological improvement.
KEYWORDS
Cerebral amyloid angiopathy, cerebral amyloid angiopathy-related inflammation, stroke, dementia, corticosteroids
INTRODUCTION
Cerebral amyloid angiopathy-related inflammation (CAA-ri) is an uncommon and aggressive subtype of CAA, which is thought to be caused by an immune reaction to deposits of β-amyloid[1]. It has a variety of presentations that can mimic other focal and diffuse neurological disorders. We describe a patient with probable CAA-ri (Table 1) mimicking stroke and review clinical, radiological and therapeutic features important for differentiating between ischaemic stroke (IS) and CAA-ri.
CASE DESCRIPTION
A 73-year-old woman who presented with an episode of generalised tonic-clonic seizure with bladder incontinence and persistent bi-temporal headache was admitted to the Accident and Emergency Department (A&E). She had no previous history of epilepsy. She was known to have cognitive impairment with memory loss, abnormal thought content and auditory-visual hallucinations for the past 4 years, and had been diagnosed presumptively with either Alzheimer’s disease or vascular dementia. She took medication to control her blood pressure (BP), and had no history of smoking, diabetes, or exposure to toxic chemicals or drugs. There was no family history of genetic diseases, cancer or immunological diseases.
Her physical examination showed no fever, BP of 102/74 mmHg, peripheral arterial oxygen saturation of 98% and a blood glucose level within the normal range. The Glasgow Coma Scale score was 15 and there were no focal neurological deficits. Respiratory and cardiovascular evaluations were unremarkable. A CT scan of the head showed left occipito-temporal cortico-subcortical hypoattenuation with sulcal effacement (Fig. 1). The patient was diagnosed with a subacute IS and admitted to a ward for management and aetiological work-up. Routine blood tests, including serum C-reactive protein level, erythrocyte sedimentation rate, glycated haemoglobin, lipid panel, creatinine, urea, liver enzymes, serum electrolytes, albumin, total protein, homocysteine levels and protein electrophoresis were unremarkable.
Testing for blood bacteria, namely Borrelia sp., Salmonella sp., Mycoplasma pneumoniae and Brucella sp., and viral pathogens, such as human immunodeficiency virus, hepatitis C virus, hepatitis B virus, Epstein-Barr virus, herpes simplex 1 and 2, cytomegalovirus and varicella-zoster virus, excluded acute infection. An electrocardiogram revealed sinus rhythm. Transthoracic echocardiography and neck ultrasound were negative for heart and vessel disease, respectively. A full body computed tomography scan showed no evidence of neoplasm. Electroencephalography revealed no epileptiform discharges.
Autoimmune tests (antinuclear antibodies, anti-neutrophil antibodies, anti-cyclic citrullinated peptide antibody, rheumatoid factor, antineuronal antibodies, antibody to extractable nuclear antigens) were negative, except for IgG anticardiolipin antibody (122 GPL; normal <20). Cerebrospinal fluid (CSF) analysis showed raised protein (0.65 g/l; normal 0.15–0.45 g/l), raised glucose content (115 mg/dl; normal 60–70% of plasma glucose, in this case normal at 72.6–88.9 mg/dl) and no cells. Cerebrospinal fluid analysis was negative for viral and bacterial pathogens, including for varicella-zoster virus, cytomegalovirus, Epstein-Barr virus, herpes simplex 1 and 2, Borrelia burgdorferi, Mycobacterium tuberculosis, Mycoplasma pneumoniae, Listeria sp., Streptococcus pneumoniae and syphilis screening. T2/FLAIR-weighted magnetic resonance imaging (MRI) sequences of the brain demonstrated asymmetric, predominantly left subcortical occipital hyperintense foci, while T1 and diffusion-weighted (DWI) images showed hypointense foci, suggestive of vasogenic oedema associated with multiple small microhaemorrhagic foci (Fig. 2).
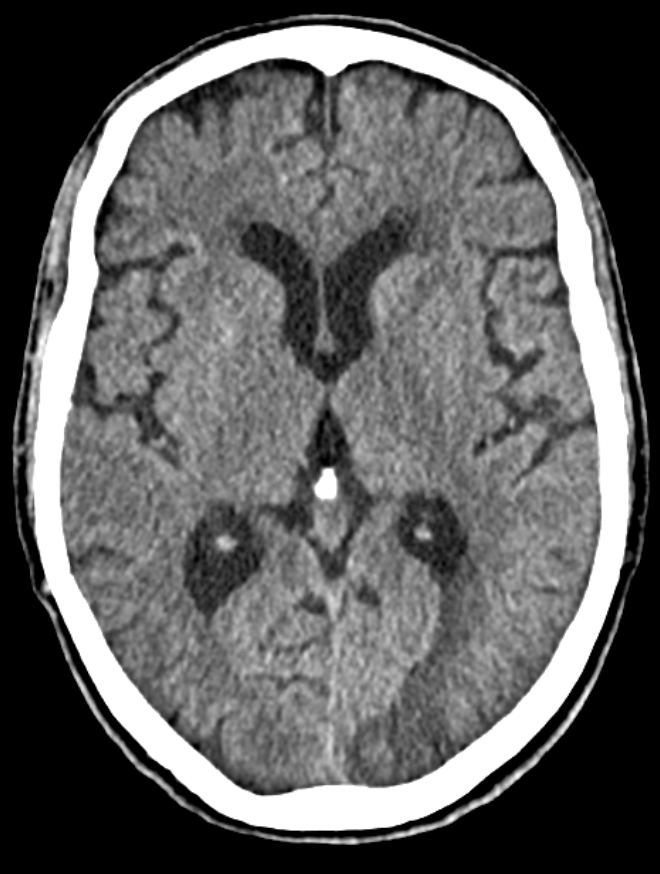
Figure 1 (click to enlarge)
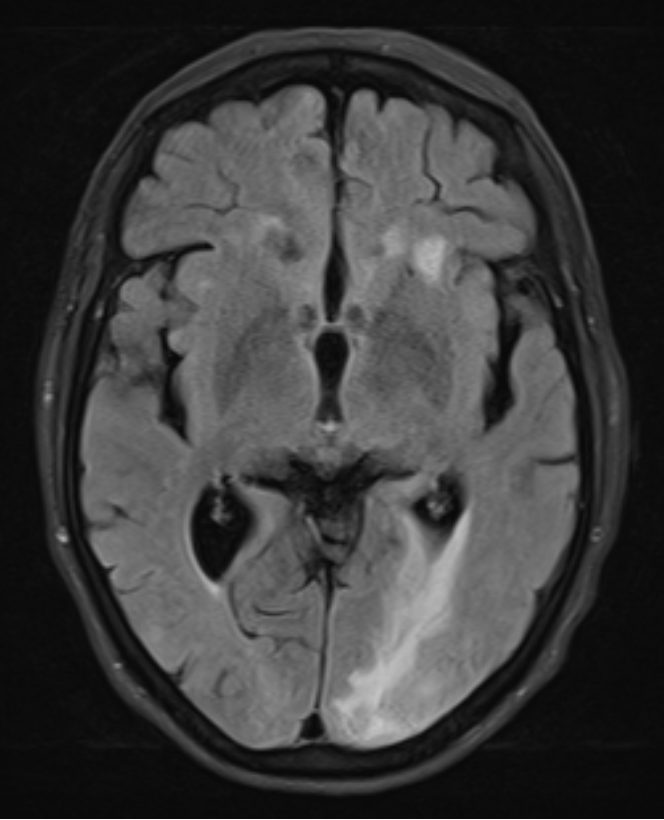
Figure 2 (click to enlarge)
Figure 1. Axial CT scan of the head showing left occipito-temporal cortico-subcortical hypoattenuation with sulcal effacement
Figure 2. Axial T2/FLAIR-weighted MRI sequences of the brain showing asymmetric, left subcortical occipital hyperintense foci
Based on these findings, and according to validated criteria (Table 1)[2], we made a diagnosis of probable CAA-ri. Levetiracetam and corticosteroid therapy were started. We first treated the patient with methylprednisolone 1000 mg/day (iv) for 5 days (after exclusion of latent tuberculosis), and then prednisolone 1 mg/kg/day (oral). The patient was discharged on a steroid tapering regime. Ten days after discharge, the patient had an episode of corticoid-induced psychosis and was reassessed in the A&E. Blood tests were normal. A CT scan of the head showed considerable regression of the previously described left occipital lesion, with concomitant reduction of local mass effect, greater individualization of cortical sulci and less deformity of the occipital marginal body, with no evidence of new cerebral parenchymal lesions (Fig. 3). Corticosteroids were reduced and neuroleptic medication introduced. Over the next 2 months, the patient remained seizure free and did not develop any new neurological deficits. Neuropsychiatric reassessment at that time demonstrated significant improvement over her previous status.
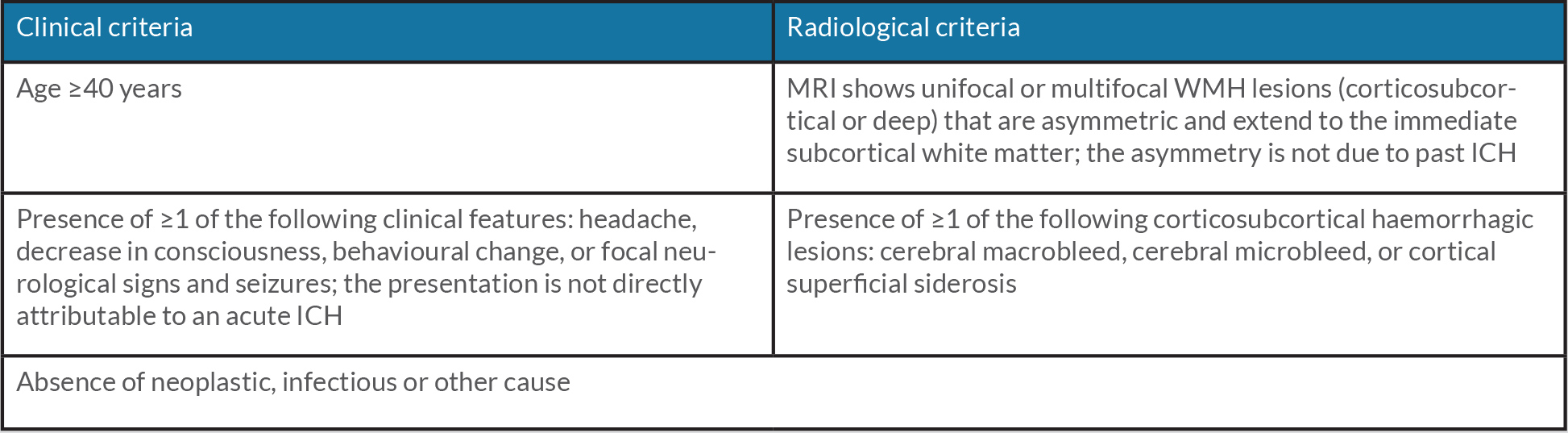
Table 1 (click to enlarge)
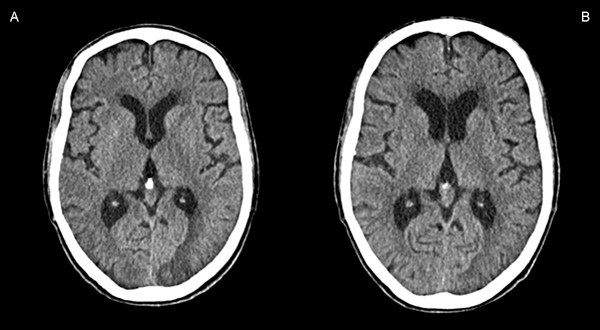
Figure 3 (click to enlarge)
Table 1. Criteria for the diagnosis of probable CAA-ri . All criteria are required for diagnosis.
CAA-ri, cerebral amyloid angiopathy-related inflammation; ICH, intracerebral haemorrhage; MRI, magnetic resonance imaging; WMH, white matter hyperintensity.
Figure 3. Comparison of axial CT scans of the head before (A) and after (B) 10 days of steroid therapy
DISCUSSION
CAA-ri is a potentially treatable disease, but its diagnosis is challenging given its clinical heterogeneity. The differential diagnosis includes infection, malignancy and autoimmune encephalitis, all excluded in this case by the diagnostic investigation described above. Another possible diagnosis is posterior reversible encephalopathy syndrome (PRES), which may be initially suspected when alterations in consciousness and motor deficits occur along with reversible subcortical vasogenic oedema on MRI. The syndrome typically presents with headache, seizures, visual impairment, focal neurological deficits and altered mental state[3]. However, PRES is accompanied by elevated BP, making the hypothesis unlikely in our case. Even though 46% of patients may present with stroke-like signs such as focal neurological signs or seizure[1], acute IS is far more common than CAA-ri. Thus, CAA-ri is rarely discussed as an alternative diagnosis in clinical practice. Nevertheless, in a case of acute onset of neurological focal signs in a patient with poor vascular risk factors and/or without evident cardiovascular disease, the differential diagnosis should include IS and CAA-ri.
As a non-invasive method, MRI is the most important examination in suspected cases of CAA-ri. Amyloid deposition and related inflammatory factors within the vascular walls contribute to hypoperfusion to vulnerable areas of white matter and lead to imaging changes on MRI, namely patchy or confluent symmetric or asymmetric areas of T2 hyperintense lesions (79.5% of cases)[2]. Clinical and radiological features allow for a probable-CAA-ri diagnosis (Table 1)[2]. Although not required by these criteria, the diagnosis can also be suggested by cerebrospinal fluid abnormalities, such as mildly elevated protein, as found in this case. Another finding that retrospectively supports the diagnosis of CAA-ri is response to immunosuppressive therapy[2]. Brain biopsy can be reconsidered in patients with probable CAA-ri who fail to respond to empiric high-dose corticosteroid therapy within 3 weeks[4].
No standard treatment regimens have been established to date for CAA-ri. However, the presented data support the use of empirical immunosuppressive therapy for patients who meet the criteria proposed for probable CAA-ri[4–6]. The primary treatment for our patient was methylprednisolone 1000 mg/day (iv) for 5 days and then prednisolone 1 mg/kg/day (oral), followed by a tapering regime. Indeed, in the vast majority of studies, corticosteroid pulse therapy is the first choice for CAA-ri. A review of 69 patients with pathologically confirmed CAA-ri revealed that 75% were treated with corticosteroids[1]. Nineteen received additional immunosuppressive therapy including cyclophosphamide, methotrexate or mycophenolate mofetil. One patient was treated with cyclophosphamide alone[1]. A positive response to treatment and clinical improvement is seen in approximately 50% of patients, but in around 33% of patients disease progression inevitably leads to death[7]. Yanjiao et al. reported the case of a 48-year-old woman diagnosed with probable CAA-ri with significant improvement of symptoms within 3 days of starting dexamethasone 10 mg/day iv[8]. Some authors have reported a positive response to treatment with clinical and radiological improvement[4,5], with 70% and 80% of clinical recovery seen within 3 and 6 months, respectively, and 84% of full clinical and radiological recovery achieved within 1 year[5]. Coulette et al. observed that despite a favourable initial evolution after treatment, there was a 42% risk of relapse, mostly within the first year (83%)[9], while Antolini et al. report 38% of recurrences happened within 24 months[10]. Recurrence was more likely if intravenous high-dose corticosteroid pulse therapy was suddenly stopped compared with slow oral tapering[10]. In cases of recurrence, pulse therapy with corticoid is often the treatment of choice, with some studies also recommending reducing the dose of corticosteroid gradually to long-term administration of oral prednisone 5 mg/day[10]. The addition of cyclophosphamide and azathioprine could be used as salvage therapy for steroid-resistant patients[3]. Attempts have also been made to try anti-amyloid drug treatment[4]. It is difficult to make a general recommendation concerning the management of individual patients, but we believe it is reasonable to start therapy with high-dose corticosteroids. Once clinical and radiological improvement is seen, consideration can be given to introducing further immunosuppressive therapy with pulsed cyclophosphamide, methotrexate or mycophenolate mofetil. The optimal duration of therapy remains to be determined but should be based on the clinical and radiological responses[1]. CAA-ri tends to require longer courses of immunosuppression, usually 6 months. However, in a case study of a patient with systemic mycobacterial infection, the decision was made to administer a relatively shortened, 3-month course of oral prednisone taper, with a positive sustained clinicoradiographic response[6]. At the 4-month evaluation, our patient had clinical and radiographic improvement (Fig. 4), with no new neurological symptoms and improved cognitive function compared with baseline and a marked reduction in the asymmetric subcortical white matter hyperintense foci.
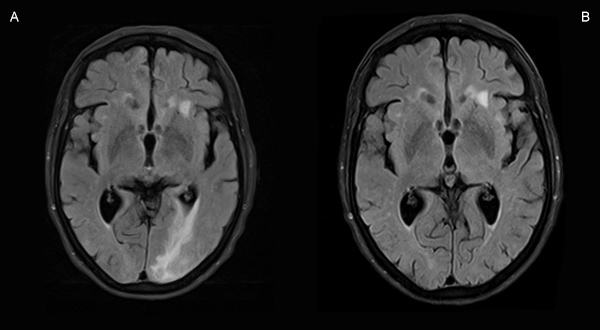
Figure 4 (click to enlarge)
Figure 4. Comparison of axial T2/FLAIR-weighted MRI sequences at diagnosis (C) and after 4 months of steroid therapy (D)
CONCLUSION
CAA-ri a rare but increasingly recognized subset of cerebral amyloid angiopathy which manifests as a reversible encephalopathy with imaging features of inflammation and oedema[1]. This case report emphasizes the need for clinicians to be aware of stroke-like presentations of CAA-ri, particularly in young patients or those without cardiovascular risk factors, since prompt management of the inflammatory process affects clinical outcomes. To improve prognosis, large sample studies to further determine better therapeutic and monitoring strategies for CAA-ri are necessary.