ABSTRACT
A 22-year-old woman presented with a 12-year history of intensifying paroxysms of anxiety, palpitations and recurrent syncope following micturition. The patient was referred to endocrinology upon discovery of hypertension. An extended family history revealed metastatic phaeochromocytoma and paraganglioma in two grand-uncles. Clinical examination revealed hypertension, with a mean 24-hour ambulatory blood pressure of 150/100 mmHg. Supine plasma normetanephrines were markedly elevated with a raised 3-methoxytyramine, while plasma metanephrines were normal. Computed tomography identified a 4.4 cm mass at the right inferolateral margin of the bladder wall. Scintigraphic imaging confirmed unifocal bladder lesion uptake with no additional metastatic lesions. Following pre-operative alpha blockade, the patient underwent a partial cystectomy. Histology confirmed a paraganglioma, and SDHB staining was lost in neoplastic cells consistent with an SDHB-related paraganglioma. Plasma normetanephrine, 3-methoxytyramine and blood pressure returned to normal postoperatively. Genetic screening identified a germline heterozygous SDHB gene variant c.723C>G. Bladder paragangliomas are a rare but important differential to consider when investigating post-micturition syncope. An extended family history should be sought and suspicion for a genetic cause should be raised, especially when the condition presents at a young age. This is the first reported case describing phaeochromocytoma or paraganglioma with the SDHB gene variant c.723C>G.
LEARNING POINTS
- Bladder paragangliomas are a rare neuroendocrine tumour which should be considered when assessing patients with haematuria and hypertension, headache, palpitations, sweating and facial pallor with micturition.
- This case highlights the importance of a thorough clinical history with an extended family history and examination in the setting of micturition syncope, which can rarely occur with bladder paraganglioma.
- Young age at presentation, a family history of phaeochromocytoma and paraganglioma (PPGL), unusual paraganglioma location, mutifocality and aggressive disease should raise the suspicion for a genetic predisposition to PPGL.
KEYWORDS
Paraganglioma, phaeochromocytoma, bladder resection, SDHB
INTRODUCTION
Micturition syncope is transient loss of consciousness before, during or after urination. There is little information in the literature as to the exact incidence of this condition, with one report citing micturition syncope in 12.2% of all cases of neurally mediated syncope [1]. It is worth noting that micturition syncope is more common in young males, thought to be related to orthostatic changes with male micturition [1].
CASE DESCRIPTION
A 22-year-old woman presented with a 12-year history of intensifying paroxysms of anxiety, palpitations and recurrent syncope after micturition. These episodes began in childhood. She described them as periods of intense anxiety with palpitations, pallor and syncope occurring during most episodes of micturition. She described immediate spontaneous recovery of symptoms following micturition. She denied haematuria. The patient presented to her general practitioner and was referred to young-adult mental health services for these symptoms, which were attributed to generalized anxiety disorder. Her blood pressure had not been assessed in childhood. However, once hypertension was discovered in early adulthood, she was referred to endocrinology for further assessment. Her extended family history revealed metastatic phaeochromocytoma and paraganglioma (PPGL) in two grand-uncles. Initially, the family history was not apparent, as the patient herself was unaware of the medical history of her grand-uncles. However, discussion with the wider family uncovered the family history, which further corroborated the evidence of a genetic predisposition to aggressive PPGL. Clinical examination was unremarkable with the exception of hypertension with a mean 24-hour ambulatory blood pressure of 150/100 mmHg.
There was marked elevation in supine plasma normetanephrines at 8874 pmol/l (reference range 0–1180 pmol/l) and 3-methoxytyramine at 198 pmol/l (reference range 0–180 pmol/l). Plasma metanephrines were normal at 110 pmol/l (reference range 0–510 pmol/l).
Computed tomography identified a 4.4 cm mass at the right inferolateral margin of the bladder (Fig. 1A). Given the young age of the patient, location of the lesion and the strong family history of PPGL, there was high clinical suspicion for a genetic cause and functional imaging was sought to rule out metastatic or multifocal disease pre-operatively. Scintigraphic imaging using I-131-labelled metaiodobenzylguanidine (131I-MIBG) showed uptake consistent with paraganglioma in the urinary bladder with no metastatic or multifocal disease (Fig. 1B).
Pre-operative alpha blockade was commenced in the week prior to planned surgery, with up-titration of phenoxybenzamine to 20 mg four times daily. The patient underwent partial cystectomy with resection of the lesion, and partial resection and repair of the anterior bladder wall. Histology confirmed a paraganglioma (Fig. 2) with positive immunostaining for synaptophysin, CD56 and chromogranin. SDHB staining was lost in neoplastic cells consistent with an SDHB-related paraganglioma. Genetic screening identified a germline heterozygous SDHB gene variant c.723C>G.
Postoperatively, blood pressure, plasma normetanephrines and 3-methoxytyramine returned to normal, and symptoms of anxiety improved. Postoperative imaging with neck-to-pelvis MRI did not show any recurrence or metastatic disease at 6 months.
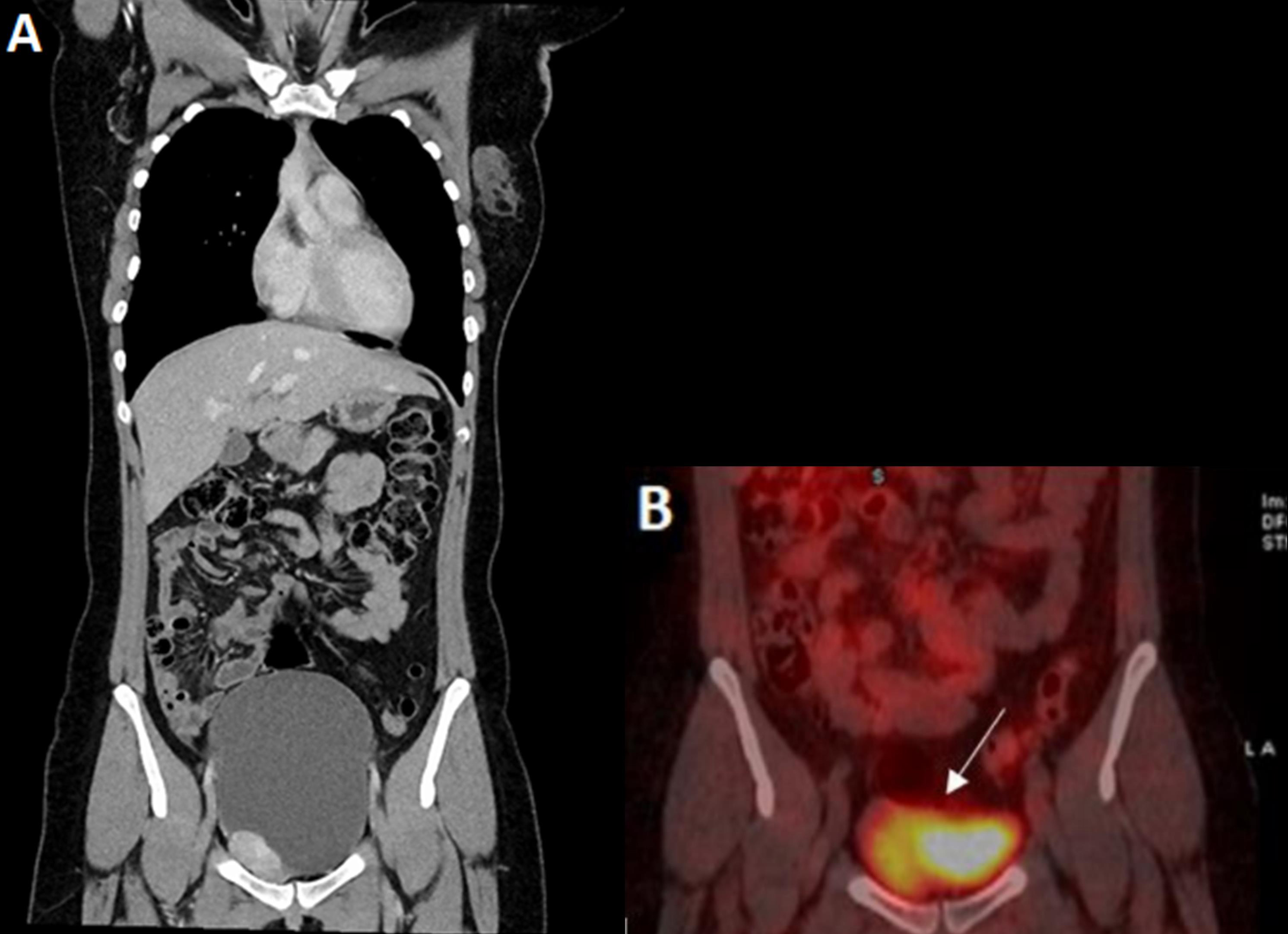
Figure 1 (click to enlarge)
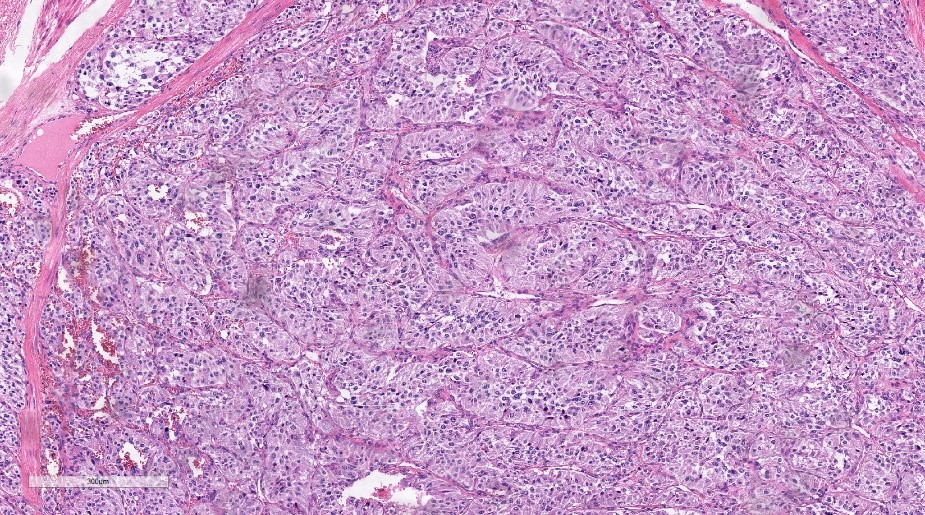
Figure 2 (click to enlarge)
Figure 1. (A) CT image showing an inferolateral bladder wall paraganglioma. (B) 131I-MIBG imaging showing radiotracer uptake at the lesion site
Figure 2. Paraganglioma histological specimen
DISCUSSION
AParagangliomas (PGL) are rare neuroendocrine tumours that are indistinguishable histologically from adrenal gland phaeochromocytomas but, in contrast, arise from extra-adrenal chromaffin cells along the sympathetic and parasympathetic nervous system [2]. Those of parasympathetic origin occur in the head and neck and are typically non-secretory, whereas sympathetic origin paragangliomas commonly secrete catecholamines and occur in the sympathetic paravertebral ganglia of the thorax, abdomen and pelvis [2]. Sympathetic paragangliomas can involve solid organs, such as the urinary bladder, and are thought to arise from chromaffin cells of the sympathetic innervation of the bladder wall [3].
In the largest case series, paragangliomas of the urinary bladder accounted for only 0.6% of all paraganglioma cases [4]. Urinary bladder paragangliomas exhibit a three-fold female preponderance with an age distribution in women of 20–30 years of age and in men of over 50 years of age [5]. They are associated with a classic triad of hypertension, haematuria and onset of symptoms of catecholamine excess with micturition, described as ‘micturition attacks’, similar to the case described above [5]. As well as micturition, other activities such as overdistension of the bladder, defecation, bladder instrumentation and sexual activity may provoke symptoms [3]. A recent multicentre study of 110 cases of urinary bladder PGL reported symptoms of catecholamine excess as the mode of discovery of the lesion in only 30% of cases. In this series, urinary symptoms of haematuria and pain on urination were more common than symptoms of catecholamine excess such as syncope, anxiety or pallor, which were reported in only 49 cases. The classic ‘micturition attack’ occurred in only 19 cases [6]. Micturition syncope is a rare presentation of urinary bladder paraganglioma. Common symptoms include hypertension, headache, palpitations, sweating and facial pallor with micturition and should alert the physician to suspect paraganglioma [4].
The diagnostic approach for PPGL relies on biochemical evidence of catecholamine excess and radiological localization [4]. Biochemical evidence of catecholamine excess can be determined by measuring plasma metanephrines (metanephrines, normetanephrines and 3-methoxytyramine) or urinary catecholamines (epinephrine, norepinephrine and dopamine) [2]. Although all values above the reference range should be further investigated, a value three times the upper limit of normal is almost always diagnostic of phaeochromocytoma or paraganglioma [2].
The Endocrine Society recommend imaging with computed tomography (CT), in the first instance, to be reserved for patients with biochemical evidence of disease [2]. PPGL have a high density, exhibiting Hounsfield units greater than 10 on non-contrast CT imaging [2]. Functional imaging is recommended when metastatic disease is suspected, when radionuclide therapy is planned or when there is an increased risk for metastatic or multifocal disease [2].
The genetic landscape of PPGL has evolved over recent years with over 40% of PPGL attributable to recognised genetic mutations [7]. More than 15 genes have been implicated in familial autosomal dominant PPGL to date, most commonly germline mutations in succinate dehydrogenase (SDH) genes followed by the familial syndromes von Hippel Lindau (VHL), neurofibromatosis type 1 (NF1) and multiple endocrine neoplasia type 2 (MEN2) [7]. The SDH gene mutations can be divided genotypically and phenotypically into four distinct categories: SDHA, SDHB, SDHC and SDHD gene mutations [7]. Loss-of-function mutations in these genes disrupt the citric acid cycle leading to accumulation of succinate and fumarate oncometabolites, which drive tumourigenesis via hypermethylation and pseudohypoxia [7]. SDHB mutations are associated with aggressive paragangliomas, which are multifocal in up to 20% of cases and confer the greatest risk of metastatic disease and mortality when compared with non-SDHB mutations [7].
Individuals with SDHB mutations should undergo lifelong follow-up with yearly biochemical and clinical screening, abdominal CT imaging every 12–24 months, and 3-yearly neck-to-pelvis MRI [7]. Following genetic counselling, cascade testing should be offered to relatives of the index case [7].
This is the first reported case describing PPGL with the SDHB gene variant c.723C>G and highlights how understanding of the genetic basis of PPGL has dramatically changed the diagnostic and long-term therapeutic approach to PPGL.