ABSTRACT
A 28-year-old female patient was hospitalized for mild–moderate hypokalaemia which was persistent despite discontinuation of beta-2 agonist bronchodilator treatment. Her past medical history was relevant for two episodes of severe hypokalaemia after active inhaled beta-2 agonist treatment for asthma crisis. Investigations revealed increased potassium in spot urine with a transtubular potassium gradient <4. A 24-hour urine analysis showed hypophosphaturia, hypocalciuria, hypomagnesuria and normal urine prostaglandins in favour of Gitelman syndrome. Oral potassium supplementation was started and genetic studies were recommended.
LEARNING POINTS
- An exhaustive aetiological work-up should be performed in young patients with persistent hypokalaemia after withdrawal of bronchodilators.
- Gitelman syndrome should be suspected in any patient with unexplained hypokalaemia, metabolic alkalosis, and a normal or low blood pressure.
KEYWORDS
Hypokalemia, metabolic alcalosis, Gitelman syndrome
INTRODUCTION
Hypokalaemia is defined as the presence of plasma potassium below 3.5 mmol/l, and is considered severe when levels are <2.5 mEq/l. There are two main causes: depletion of the total potassium content of the organism, or excessive entry of potassium into cells, especially muscle cells. Special attention should be given to those cases with abnormal renal losses. Gitelman syndrome is one of the causes of hypokalaemia, with an estimated prevalence of 1:40,000 [1, 2].
Serum potassium between 3 and 3.5 mmol/l rarely causes symptoms; however, levels below 3 mmol/l often result in muscle weakness, cramps, fasciculations, paralytic ileus, hypoventilation, arterial hypotension and in severe cases, rhabdomyolysis, tetany, paralysis and respiratory failure. Persistent hypokalaemia may compromise renal concentrating ability and cause polyuria with secondary polydipsia [2, 3].
CASE DESCRIPTION
A 28-year-old female patient was hospitalized for mild–moderate hypokalaemia which was persistent despite discontinuation of bronchodilator treatment. Her medical history was relevant for bronchial asthma treated with beta-2 agonist inhalers and two previous episodes of tetany due to severe hypokalaemia after intensive treatment with salbutamol (2011) and terbutaline (2017) for asthma crisis. The patient referred occasional episodes of paraesthesia, fatigue and cramps, and she had stopped regular sports activities due to the rapid onset of fatigue. There was no history of vomiting, diarrhoea, drugs (especially no diuretics, laxatives, gentamicin or colistin), salt craving or polydipsia. There was no relevant family history. Her treatment included potassium 600 mg (8 mEq) three times per day, and inhaled anticholinergics. She did not smoke or take drugs and had no drug allergies.
On physical examination, her height was 173 cm, weight was 65 kg, blood pressure was 126/83 mmHg and heart rate was 83 bpm, she was afebrile, and oxygen saturation was 98% on room air. Mucous membranes were moist and well-coloured. No skin lesions were found. Heart sounds were regular without murmurs or rubs, pulses were normal, and there was no oedema. Lungs were clear. The abdomen was tender but not distended, and without palpable masses.
Laboratory blood tests revealed hypokalaemia (3.09 mmol/l) and chloride 95 mmol/l, normal total and corrected calcium and sodium, low ¬normal magnesium, and preserved renal function. Subsequently, we assessed urinary potassium excretion to distinguish between renal and non-renal losses and also assessed acid-base status. Spot urine potassium was >20 mmol/l in two repeated analyses indicating inappropriate renal loss. Urine potassium concentration in a single urine sample can serve as an initial guideline but is greatly influenced by whether the urine is concentrated or dilute. It is therefore advisable to determine the transtubular potassium gradient (TTKG) in a 24-hour urine sample, which corrects urinary K for water reabsorption in the collecting tubule and allows the presence and magnitude of mineralocorticoid action in the distal convoluted tubule (DCT) to be assessed (Fig. 1).
Our patient was normotensive, TTKG was >7 and serum aldosterone was normal, while renin activity was elevated but with a normal ratio. Venous blood gas analysis showed metabolic alkalosis (pH 7.47, pCO2 46.4 mmHg, HCO3 32.6 mmol/l). In this setting of TTKG>7 and metabolic alkalosis, urine chloride was determined to be >20 mmol/l. As our patient had no history of vomiting, urine chloride >20 mmol/l suggested diuretic intake or Bartter/Gitelman syndrome. The patient denied diuretic intake and occult intake of thiazide or loop diuretics was ruled out by urinary analysis. In light of these results, we sought to differentiate between Bartter and Gitelman syndrome (Table 1).
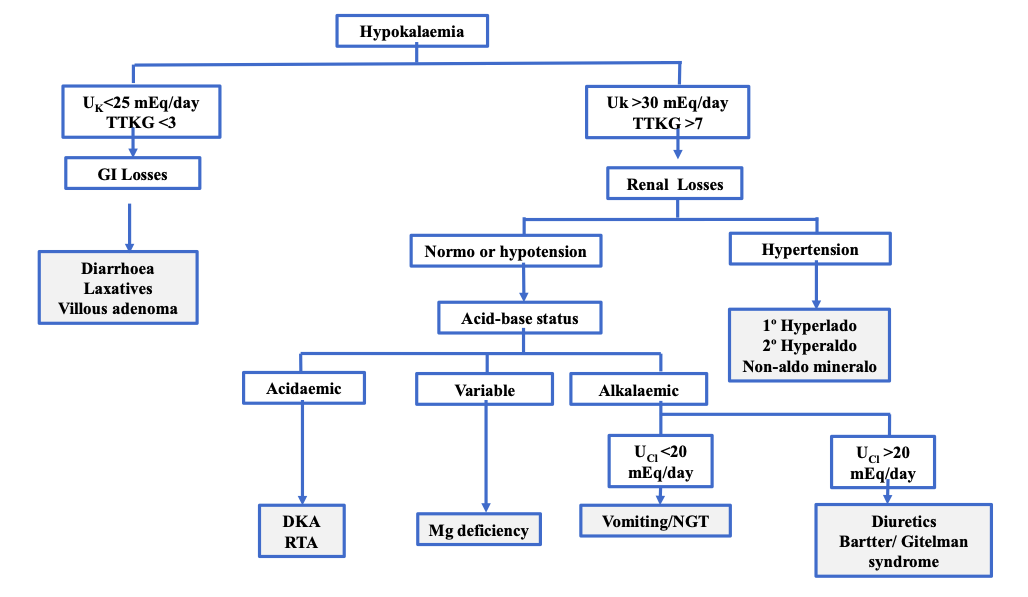
Figure 1 (click to enlarge)

Table 1 (click to enlarge)
Figure 1 Work-up of patients with hypokalaemia
Table 1 Differential diagnosis between Bartter and Gitelman syndrome
The 24-hour urine found hypophosphaturia (201.06 mg/24h), hypocalciuria (21.06 mg/24h) and hypomagnesuria (43.92 mg/24h) without other abnormalities. Subsequently, analysis of urine prostaglandin E2 came back normal. No proteins or red blood cells were found in the urine. Abdominopelvic tomography and renal ultrasound were normal.
In light of these results, we established the diagnosis of Gitelman syndrome, since the condition had presented in adulthood, and the predominant symptoms were muscle weakness, tetany and asthenia, even though hypomagnesaemia was absent. In contrast, Bartter syndrome typically manifests in childhood, is associated with growth retardation and the main clinical presentation is loss of salt and water rather than muscle weakness. Genetic studies were requested. Two variants of uncertain significance in the SLC12A3 gene associated with Gitelman syndrome were found, which could explain the patient's phenotype. The patient was treated with oral magnesium (even if levels were normal) and began oral potassium.
DISCUSSION
In the case, the patient presented severe hypokalaemia after beta-2 agonist treatment. The most important metabolic side-effect of beta-2 agonist use is hypokalaemia.
Hypokalaemia is thought to be related to the stimulation of beta-2 receptors bound to the Na/K ATPase system in skeletal muscle, which leads to an influx of potassium into the cells, rather than to other mechanisms such as insulin release. This effect is especially important when beta-2 agonists are used intravenously and occurs less frequently with inhalation, as in the case presented, when substitution treatment is rarely required [4]. This fact led to the performance of other studies in our patient.
Work-up for hypokalaemia includes assessment of urinary potassium excretion to distinguish between renal potassium losses from other causes of hypokalaemia (gastrointestinal losses, transcellular potassium shifts) and the assessment of acid-base status, since some causes of hypokalaemia are associated with metabolic alkalosis or metabolic acidosis. Metabolic alkalosis with potassium wasting and hypertension occurs with primary mineralocorticoid excess or a disorder that mimics a mineralocorticoid excess state, renovascular disease, or the surreptitious ingestion of diuretics by a patient with underlying hypertension. Blood pressure in our patient was repeatedly normal and hormone studies did not suggest a mineralocorticoid excess state. In patients with metabolic alkalosis with renal potassium wasting and a normal blood pressure, hypokalaemia is most often due to diuretic use, vomiting, or Gitelman or Bartter syndrome.
The measurement of urinary chloride is important in determining the cause as levels <20 mmol/l suggest diuretic use (unlikely), cystic fibrosis, vomiting, nasogastric tube suction or chloridorrhea, while levels >20 mmol/l suggest recent diuretic intake in the setting of primary hyperaldosteronism, Cushing’s disease, ectopic ACTH production, Liddle syndrome, exogenous mineralocorticoids production in hypertensive patients, and Batter/Gitelman syndrome in normotensive/hypotensive patients. Urine chloride was >20 mmol/l, the patient did not have a history of vomiting, diarrhoea or diuretic intake and blood pressure was normal. Consequently, a tubulopathy was considered.
Hypokalaemic salt wasting tubulopathies are a heterogeneous group with common features: recessive autonomic inheritance, renal salt wasting, metabolic alkalosis, and increased renin and aldosterone levels with normotension. There are three well-differentiated entities within the salt-losing tubulopathies: prenatal Bartter syndrome, classic Bartter syndrome and Gitelman syndrome [1, 2]. From a clinical point of view, Bartter syndrome is detected in childhood with growth retardation, polyuria, polydipsia and anorexia. Gitelman syndrome has a milder phenotype and later presentation and differs from Bartter syndrome in that patients have hypomagnesaemia and hypocalciuria. They may be asymptomatic or present with transient muscle weakness, paraesthesias, paralysis and even rhythm disturbances. Bartter syndrome type 3 and Gitelman syndrome usually present in adolescence or early adulthood and in the absence of genetic confirmation, it can be difficult to differentiate between the two disorders. Type 3 is caused by a CLCNKB gene mutation and can be clinically indistinguishable from Gitelman syndrome. Mutations that usually cause Gitelman syndrome are much more common than those causing Bartter syndrome type 3. In support of the diagnosis in our patient, she was 28 years old, her blood pressure was normal, she reported muscle weakness, paraesthesias and fatigue, and laboratory analysis showed hypocalciuria with normal urine prostaglandin levels.
From a pathogenetic point of view, in prenatal Bartter, mutations have been described in the NKCC2 receptor (Na, K, Cl transporter) and ROMK (outflow transporter of K from the tubular cells of the ascending loop of Henle), in classic Bartter, the mutation lies in the receptor of the basolateral ClC-Kb channel that allows Cl to exit the cell [1, 2], and in Gitelman syndrome, the defect lies in the NCCT channel, responsible for Na and Cl reabsorption from the DCT (encoded on chromosome 16q13). However, there is no phenotype–genotype relationship and phenotypic transitions from one syndrome to another have been described.
Although the majority of mutations in Gitelman syndrome patients were found in SLC12A3, in a small percentage of subjects mutations in the CLCNKB gene, coding for the chloride channel Kb, were demonstrated to cause the disease. In addition, a relevant percentage of Gitelman syndrome clinical cases are heterozygous carriers or do not show any mutations in SLC12A3 [2, 3, 5] and perhaps this explains the wide range of variability in clinical presentation, and the presence of alternative routes for chloride to exit the DCT cell have been evoked.
Adequate diagnosis and treatment are important, since prolonged hypokalaemia can lead to renal damage including interstitial nephritis, tubular atrophy, interstitial fibrosis and acute renal failure. Prognosis and treatment are more favourable in Gitelman syndrome.
The main goal of treatment is to correct or minimize electrolyte disorders and extracellular volume depletion. The first-line approach consists of oral supplementation with sodium chloride, potassium chloride, and when magnesium levels are reduced, magnesium salts. NSAIDs may be helpful in these patients; however, they have important gastrointestinal side effects and can diminish renal function or even precipitate severe kidney injury. In addition, angiotensin converting enzyme inhibitors reduce blood pressure, which could be problematic in patients who already have low or low-normal blood pressure, and so should generally be added when other therapeutic tools have not been effective. Genetic counselling should be offered to those individuals carrying the disease-causing mutation, informing them that there is a 25% risk of transmitting the mutation to their offspring.
CONCLUSIONS
In this case, hypokalaemia was initially assumed to be due to an inhaled beta-2 agonist but the patient actually had a hypokalaemic salt-wasting tubulopathy. Mild chronic hypokalaemia in young patients requires full attention and further investigations.