ABSTRACT
Pulmonary arterial hypertension (PAH) is an increasingly recognised clinical entity that is associated with connective tissue disease (CTD) in up to one quarter of all diagnoses. Sjögren’s syndrome (SS) is a chronic autoimmune disease characterised by ocular and oral dryness resulting from lacrimal and salivary gland dysfunction. Additionally, SS may involve virtually any organ system and, as a result, the disease is characterised by pleomorphic clinical manifestations. However, SS-PAH reports are scarce, and the area remains insufficiently studied. We present a case of a 75-year-old female with a new diagnosis of PAH and SS.
LEARNING POINTS
- SS is a chronic autoimmune disease that may involve virtually any organ system, representing a rare cause of PAH.
- The exclusion of SS as a possible diagnosis is needed before a diagnosis of idiopathic PAH can be made.
- Routine screening of PAH is recommended in SS patients and future studies should clarify the optimal management of these patients, including immunosuppressive therapy.
KEYWORDS
Primary Sjögren’s syndrome; pulmonary artery hypertension
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a classified as Group I pulmonary hypertension (PH) by the World Symposium on Pulmonary Hypertension (WSPH) [1]. While being increasingly recognised, PAH associated with connective tissue disease (CTD) corresponds to up to 25% of all diagnoses, second only to idiopathic PAH [1]. Several studies have demonstrated that CTD-PAH has a poorer prognosis than other PAH groups despite similar therapy, raising concerns on the optimal approach in this subset of patients [2,3].
Sjögren’s syndrome (SS) is a chronic autoimmune disease characterised by pleomorphic clinical manifestations, as it may involve virtually any organ system [4]. Pulmonary manifestations in patients with primary SS include a variety of interstitial lung diseases, airway disease, pleurisy, lymphoma, amyloidosis, pulmonary vasculitis, granulomatous disease and diaphragmatic myopathy [5]. Although PAH can be accompanied by primary SS, this pulmonary manifestation is rare and more frequently associated with other CTD – systemic sclerosis being the most common CTD-associated PAH in Western countries [1,6]. Considering the paucity of reports on SS-PAH, optimal treatment is yet to be determined and more studies are necessary [1].
CASE DESCRIPTION
This case report describes a 75-year-old female with previous medical history of hypertension, diabetes mellitus and atrial fibrillation. She had no personal or familial history of autoimmune diseases.
She was admitted to the emergency department (ED) with complaints of progressive, exertional dyspnoea that started six months before, associated with peripheral oedema, orthopnoea and paroxysmal nocturnal dyspnoea. Additionally, she complained of xerostomia and xerophthalmia that started twenty years ago.
At admission to the ED she was haemodynamically stable (blood pressure of 104/64 mmHg, heart rate of 86 bpm); peripheral saturation was 86% on room air and she was apyretic. Auscultation revealed diminished lung sounds at the right base, and bibasilar crackles. An erythematous, pruritic rash with annular, discoloured plaques was prominent on both forearms, limbs and torso (Fig. 1); pitting oedema up to the knees was noted. Chest radiography was consistent with right pleural effusion and laboratory results were unremarkable except for an elevated brain natriuretic peptide (BNP) of 1585 pg/mL.
She was hospitalised and started intravenous loop diuretics with progressive improvement of hypervolemia. A transthoracic echocardiogram revealed moderate dilation of the right chambers and left atrium, concentric hypertrophy of the right ventricle, an estimated pulmonary artery systolic pressure of 55+15 mm Hg and right ventricular dysfunction. A right heart catheterisation was performed and was compatible with combined postcapillary and precapillary pulmonary hypertension (mean pulmonary artery pressure, mPAP=48 mm Hg; pulmonary capillary wedge pressure, PCWP=18 mm Hg and pulmonary vascular resistance, PVR=10 Wood units).
An abdominal ultrasound, a chest computed tomography angiography and a ventilation-perfusion scan were performed and results were unremarkable. Viral serologies were negative.
A skin biopsy of the forearm rash was obtained and was compatible with granuloma annulare (GA). Autoimmune serologies were positive for anti-nuclear antibody 1/1000 (dense fine speckled) and anti-SSA/anti-Ro52. A salivary gland function scan was positive for bilateral subacute parotitis, and a salivary gland biopsy was consistent with SS (focus score 1.4/4 mm2).
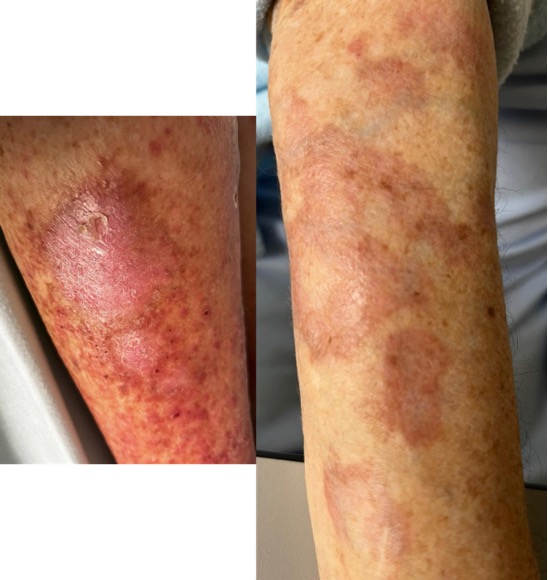
Figure 1 (click to enlarge)
Figure 1. Erythematous rash with annular, discoloured plaques in both forearms, limbs and torso, suggesting granuloma annulare.
A 6-minute walking test was performed, covering 240 metres, with a minimum peripheral oxygen saturation of 89%.
A diagnosis of SS with mucocutaneous and vascular involvement (EULAR Sjögren’s syndrome disease activity index – ESSDAI: 5 points) and high-risk combined PTH was made. The patient started prednisolone 5 mg q.i.d., sildenafil 25 mg t.i.d. and sequentially ambrisentan 5 mg q.i.d., without intercurrences. At 3 months after discharge, improvement of dyspnoea and skin lesions was observed; the New York Heart Association (NYHA) functional class was II, blood pressure 103/74 mm Hg and BNP lowered to 748 pg/mL; no further symptoms were noticed.
DISCUSSION
Primary SS is a chronic autoimmune disease, which can involve different organs and systems along with exocrine glands [7]. The association of GA with SS is rare and, to the best of our knowledge, only two cases have been reported [8,9].
PAH is a rare complication of primary SS and is associated with high morbidity and increased mortality [1]. The prevalence of this pulmonary manifestation is probably underdiagnosed; additionally, periodic risk assessment is recommended in patients with SS, as a low-risk category is associated with better long-term outcomes and could be applied as a therapy goal [1]. Additionally, as observed in this case, the development of PAH may not be parallel to disease activity and damage [1]. In patients with PAH, the presence of primary SS should be carefully investigated as it may be a non-specific and quiescent manifestation in this group of patients [1].
The therapeutic approach to this patient is challenging, considering the presence of combined PH in a patient with CTD. Despite having risk factors for PH associated with left heart disease, considering the magnitude of elevation of PVR in a patient with CTD and a high risk of death, the team decided to start sequentially a combination PAH-target therapy, with supervision during hospitalisation [10].
In the largest cohort study of SS-PAH, 84.5% received immunosuppressants and 88.3% PAH-target therapy [1]. Since PAH is one of the organ manifestations of CTD, is possible that immunological mechanisms are involved in its pathophysiology [11]. Clinicians might initially – or concomitantly to PAH-targeted therapy – administer immunosuppressants, but their prognostic impact and optimal therapy are unknown[1,12]. A retrospective cohort by Launay et al. showed possible haemodynamic improvement in a patient with SS-PAH with the use of monthly pulses of cyclophosphamide, but no impact on symptom relief or NYHA class [10]. Further research is needed to determine the best therapeutic approach in patients with SS-PAH.