ABSTRACT
We report the case of a 70-year-old man diagnosed with late-onset Wilson disease (WD) with mild neurological symptoms only and a new mutation in the ATP7B gene. A compound mutation of the ATP7B gene was found with the variant c.98T>C p(Met33Thr) in exon 2, in heterozygosis, and variant c.2224G>A (Val742Ile) in exon 8, in heterozygosis. Patient age should not be a determinant for excluding WD. Genetic sequencing is an important tool for the discovery of new genetic mutations.
LEARNING POINTS
- Wilson disease (WD) is an autosomal recessive disorder of copper metabolism
- Patient age should not exclude WD, and symptoms compatible with WD should raise suspicion for WD even in older people.
- Genetic sequencing is an important tool in the discovery of new genetic mutations.
KEYWORDS
Wilson disease, ATP7B gene, older age
BACKGROUND
Wilson disease (WD) is an autosomal recessive disorder of copper metabolism, characterized by copper accumulation in tissue, which results in hepatic, neurological and/or psychiatric symptoms. The underlying genetic defect causing WD has been identified within the ATP7B gene, mapped to chromosome 13q14.1 [1]. The 7.5 kb WD gene transcript encodes a 1465 amino acid protein of the P-type adenosine triphosphatase family which contains six copper-binding regions, an adenosine triphosphate (ATP) domain, a transmembrane cation channel, a phosphorylation region, and a transduction domain responsible for the conversion of energy of ATP hydrolysis to cation transport[2–5]. More than 600 mutations have been identified to date [6]. Most patients are compound heterozygotes, that is, they carry two different mutations [7]. Symptoms usually first appear in the second and third decades of life [8], but there are several reports of late-onset WD [9, 10].
CASE DESCRIPTION
The patient was a 70-year-old man with action tremor in the distal upper extremity, deteriorated handwriting and micrographia. Tremor was present predominantly in the right hand and emerged during the course of goal-directed activity (drinking from a glass of water or writing). The patient was a 70-year-old man with action tremor in the distal upper extremity, deteriorated handwriting and micrographia. Tremor was present predominantly in the right hand and emerged during the course of goal-directed activity (drinking from a glass of water or writing). There was no history of WD in his family and his parents were not consanguineous. We noted low levels of serum ceruloplasmin (16.3 mg/dl, RV: 18–45 mg/dl). 24-Hour urinary cooper excretion was 120 mg/24 h (normal <100 mg/24 h). Anterior segment examination of the eyes confirmed the findings of early Kayser-Fleischer rings (Fig. 1).

Figure 1 (click to enlarge)
Figure 1. Early Kayser-Fleischer rings on slit-lamp examination
There were copper deposits in an arcuate pattern in the superior and inferior aspect of the cornea and copper deposits on the anterior surface of the lens. There was no liver disease. Brain MRI showed no copper deposits in the basal ganglia. Molecular genetic analyses of the ATP7B gene were performed with informed consent of the patient. Genomic DNA was extracted from whole blood using QIAsymphony. The first library was prepared by conventional PCR followed by Nextera (Illumina) and subjected to second generation sequencing (Illumina). The variant c.98T>C p(Met33Thr) was found in exon 2 of the ATP7B gene, in heterozygosis. This variant substituted a methionine for a threonine at codon 33 of the translated protein, considered a rare variant (gnomAD: 0.06%), and was described as a variant of uncertain meaning in the ClinVar database (rs184868522). Variant c.2224G>A (Val742Ile) was found in exon 8 of the ATP7B gene in heterozygosis. That variant substituted a valine for an isoleucine at codon 742 of the translated protein, is a novel variant, is absent in population databases consulted, and has not been described in individuals with ATP7B-associated disease. Our patient met the diagnostic criteria for WD given in the EASL Clinical Practice Guidelines. Treatment with zinc sulfate was started at a dose of 100 mg per day. Urinary copper initially decreased but rose again after 8 months (Fig. 2). The dose of zinc sulfate was increased to 150 mg and the low-copper diet was adjusted. There was a sustained fall in urinary copper (Fig. 2) and a reduction in Kayser-Fleischer rings evaluated after 1 year.
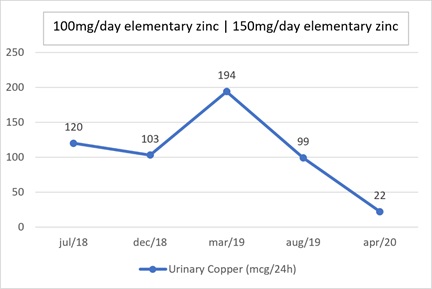
Figure 2 (click to enlarge)
Figure 2. Urinary copper timeline during zinc treatment
DISCUSSION
We report the case of an elderly man diagnosed with late-onset WD with mild neurological symptoms only and a new mutation in the ATP7B gene. We attribute the mild presentation of the disease to a missense-type mutation leading to low penetrance. We opted for treatment with zinc sulfate only due to the mild presentation and the risk that treatment with d-penicillamine and trientine could cause neurological worsening.
Severe impairment of ATP7B function leads to an aggressive phenotype and earlier onset of the disease. However, mild impairment of ATP7B leads to late onset of disease. It is possible that insertion/deletion and nonsense mutations alter the reading frame and result in truncation of the protein, thus causing more severe and earlier disease. On the other hand, missense mutations likely lead to mild phenotypes [5, 11]. Despite the biological rationale that the type of mutation in the DNA leads to a deformation in the protein, this observation is not linear in WD. In some studies, age at WD onset and type of clinical presentation did not show a significant correlation with ATP7B genotype [12, 13]. The inaccurate genotype-phenotype relationship must be influenced by modifier genes and epigenetic regulation of gene expression which are not yet fully understood [14].
In a Brazilian population, a first report determined that the c.3402delC mutation at exon 15 was the most common, with an allelic frequency of 30.8%. The second most frequent mutation was the c.2123T>C at exon 8, with an allelic frequency of 14.1% [15]. But in a second report, the c.3207C>A substitution at exon 14 was the most common mutation (allelic frequency 37.1%) followed by the c.3402delC at exon 15 (allelic frequency 11.4%) [16]. Almost half of all documented Brazilian mutations of the ATP7B gene were located in exons 14 and 15 [15, 16]. In our case, a compound heterozygous mutation was detected in exon 2 and exon 8. This exon 8 mutation is being reported for the first time. Genetic sequencing is an important tool for the discovery of new genetic mutations.
Patient age should not exclude WD. Although the majority of WD patients present symptoms between the ages of 5 and 35 [7], the disease has been reported in patients as young as 3 [17] and as old as 84 [9]. Indeed, about 4% of patients present beyond the fourth decade [18]. Features compatible with WD should raise suspicion for the diagnosis even in older patients [10].