ABSTRACT
A 60-year-old man, with a history of familial lipodystrophy, hypertriglyceridaemia, hepatic steatosis and bone cysts, was admitted due an acute coronary event. Coronary angiography showed significant stenosis in the left anterior descending artery, which was treated. Transthoracic echocardiography showed a slightly dilated left ventricle with diffuse and heterogeneous thickening of its walls, slightly decreased left ventricular function and reduced global longitudinal strain. Due to these echocardiographic findings, cardiac magnetic resonance imaging was requested, which identified intramyocardial diffuse fibrosis of the basal septum and points of insertion of the left and right ventricles, without oedema, microvascular obstruction or myocardial infarction. Owing to the constellation of symptoms and distinctive features on cardiac imaging, a diagnosis of Berardinelli-Seip congenital lipodystrophy (BSCL) was suspected, which was confirmed through genetic testing of the pathogenic variants in BSCL2 and AGPAT2. BSCL is a rare autosomal recessive syndrome characterized by the congenital absence of adipose tissue and triglyceride deposition in other tissues, such as muscle, liver and heart.
LEARNING POINTS
- Berardinelli-Seip congenital lipodystrophy (BSCL) is a rare congenital lipodystrophy, with an incidence of 1–9 per million population, which is usually diagnosed at birth and is associated with pathogenic variants of the BSCL2 and AGPAT2 genes.
- Due to the absence of functional adipocytes, lipid storage occurs in other tissues, including skeletal muscle and liver.
- Diagnosis is based on the presence of three major or two major and two minor characteristics.
KEYWORDS
Berardinelli-Seip, myocardial infarction, familial lipodystrophy, thrombocytosis, triglycerides
CASE DESCRIPTION
We present the case of a 60-year-old man, a former smoker, with previously diagnosed arterial hypertension, dyslipidaemia (particularly hypertriglyceridaemia), thrombocytosis, a history of familial lipodystrophy, hepatic steatosis and a bone cyst on the right arm with previous fracture in 2019. He was admitted to hospital with anterior wall ST segment elevation myocardial infarction. Dual antiplatelet treatment was administered and the patient underwent emergent cardiac catheterization, showing significant stenosis in the left anterior descending artery (LAD), which was successfully treated with a stent. Serological tests (HIV, HBV and HCV) were negative.
During hospitalization, the patient presented Killip class I and a maximum troponin I level of 1398 ng/l, without recurrence of thoracic pain or arrhythmic events. A transthoracic echocardiogram was performed, showing a slightly dilated left ventricle with diffuse and heterogeneous thickening of its walls, slightly decreased left ventricular ejection fraction (LVEF 51%), reduced global longitudinal strain (GLS –13.3%) and diastolic dysfunction (E/Eʹ of 13) (Fig. 1). The papillary muscles were thickened but normally inserted. No asymmetries in segmental contractility, valvular dysfunction or compromise of the right ventricle were described. The right ventricle presented normal thickening.
Due to the echocardiographic features, hypertriglyceridaemia of 1012 mg/dl and the patient’s family history of lipodystrophy, the possibility of Berardinelli-Seip congenital lipodystrophy (BSCL) was considered. The patient presented with the major characteristics of lipoatrophy (highly developed musculature for the amount of exercise practiced), hypertriglyceridaemia and hepatic steatosis, and the minor characteristics of hypertrophic cardiomyopathy and bone cysts (Table 1). Hyperglycaemia, frequently associated with this condition, was not documented during this hospitalization.
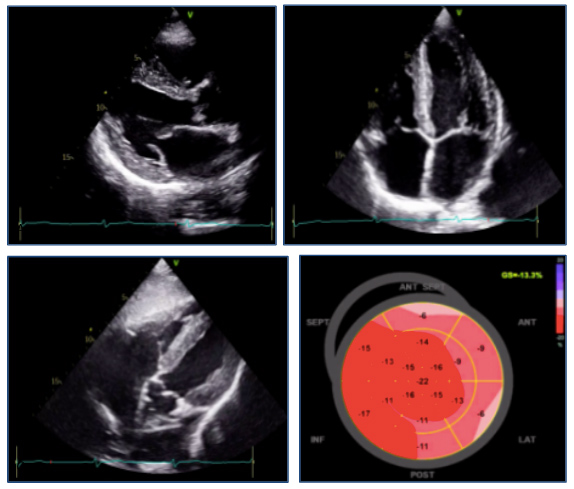
Figure 1 (click to enlarge)
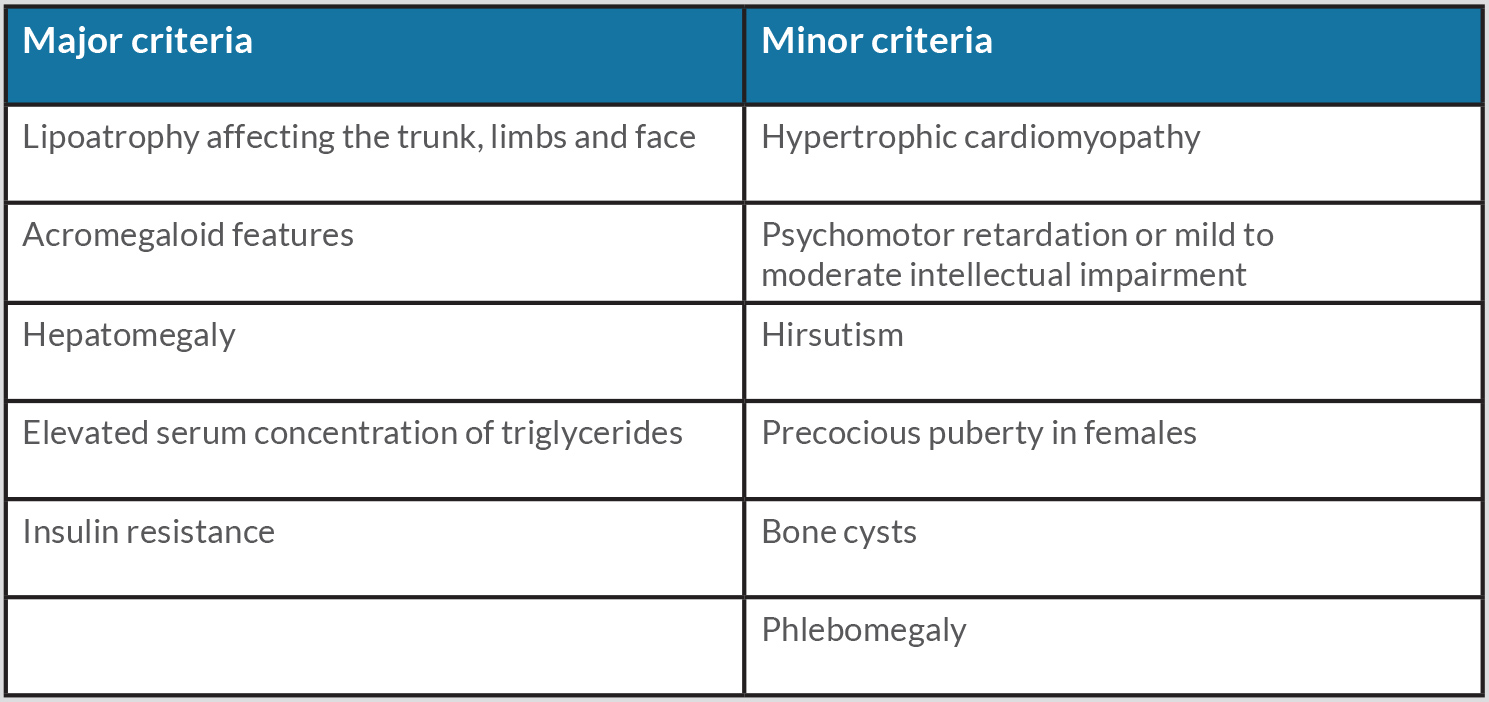
Table 1 (click to enlarge)
Figure 1. Echocardiography images: parasternal (upper left panel), apical four-chamber (upper right panel) and subcostal views (lower left panel), and global longitudinal strain (lower right panel)
Table 1. Major and minor criteria for the diagnosis of Berardinelli-Seip syndrome
To further study the possibility of BSCL, cMRI was performed and showed a hypertrophic and slightly dilated left ventricle with diminished LVEF. Intra-myocardial fibrosis was particularly marked on the basal septum and points of insertion of the left and right ventricles, with diffuse fibrosis of the medial apical segment of the septum and anterior wall. No areas of myocardial oedema, microvascular obstruction or myocardial infarction were noted (Fig. 2). The absence of cutaneous adipose tissue with hepatic steatosis on cMRI, in addition to the features described, supported the diagnosis of congenital lipodystrophy, particularly BSCL.
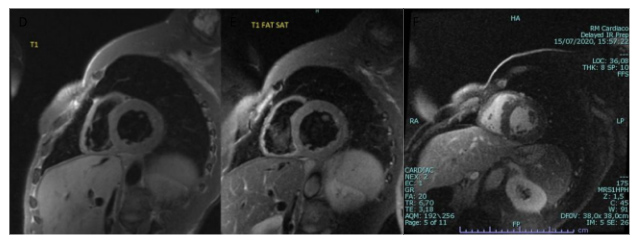
Figure 2 (click to enlarge)
Figure 2. Magnetic resonance images: short axis T1-weighted (left panel), short axis T1-weighted with fat suppression (middle panel), and short axis late gadolinium enhancement with myocardial fibrosis at the insertion of the left and right ventricles (right panel)
The patient initiated statin and beta-blocker therapy with a good response. Subsequently, the diagnosis of BSCL was confirmed with positive identification of a heterozygotic mutation in the AGPAT2 gene.
Lipodystrophy syndrome can be acquired or congenital. The fact that our patient had the AGPAT2 gene mutation and a recently discovered familial history, suggests he had the congenital form.
The patient remains stable and well controlled under lipid-lowering and dietary measures with decreasing levels of triglycerides, as well as treatment with beta-blockers and double antiplatelet therapy. Regarding thrombocytosis, the patient underwent JAK2 analysis, which was negative, and was referred to Haematology for further study. To the best of our knowledge, there is only one reported case of an association between BSCL and essential thrombocytosis [1].
BSCL has an autosomal recessive mode of transmission, so all of the patient’s children were tested, but none were positive for the disease.
DISCUSSION
BSCL is usually identified during the neonatal period, but in this patient the absence of insulin resistance and of some of the phenotypic characteristics, namely acromegaly and acanthosis nigricans, probably masked the other signs and symptoms; a diagnosis was only made due to an acute event. There are no curative options, but lifestyle modifications and conventional antihyperglycaemic and lipid-lowering medications are used. Insulin, sulfonylureas, metformin and pioglitazone may be employed to manage hyperglycaemia. Treatment with fibrates and statins may be successful for hypertriglyceridaemia, although a more aggressive approach, namely plasmapheresis, may sometimes be needed to prevent pancreatitis in patients with triglyceride counts persistently above 1000 mg/dl despite therapy [2]. Since 2014, recombinant leptin therapy has been approved to treat patients with severe insulin resistance and steatohepatitis [3]. Regarding cardiac compromise in BSCL with AGPAT2 mutation, several alterations have been described, but the underlying pathophysiology has not been completely elucidated. Early atherosclerosis has a prevalence of 60% and manifests before the age of 45. In a review of the literature, almost all patients presented with cardiomegaly and with left ventricular hypertrophy, ranging from mild to severe, with concentric hypertrophy being the most common type. Furthermore, in some patients, this may progress to left ventricular systolic dysfunction, but information is lacking on diastolic function and arrhythmias [4, 5].
CONCLUSION
The role of IgA is not limited to antibacterial and antiviral functions in mucosal immunity. Serum IgA can reduce cytokines and leucocyte, monocyte and neutrophil chemotaxis and send inhibitory signals to antigen-presenting cells. It could have an anti-inflammatory role or be a protective mechanism against tissue damage. Serum IgA levels may be helpful to identify patients not responding to traditional therapy.
Since the disease is rare, retrospective or prospective multicentre studies are required to determine if IgA could be a disease activity biomarker. It would be interesting to further investigate IgA function in Takayasu vasculitis pathophysiology.
Life expectancy is significantly reduced by 30 or more years, with cause of death being mostly due to infections, liver disease, renal failure, acute pancreatitis or myocardial infarction.