ABSTRACT
Takayasu arteritis is a systemic vasculitis of the large vessels and mainly affects Japanese and Southeast Asian women in the second and third decades of life. Inflammatory infiltrate affects the full thickness of the vessel wall, inducing progressive lumen stenosis and occlusion. The main biomarkers of disease activity are the ESR, CRP and serum levels of circulating cytokines.
This case report describes the clinical history of a young woman with Takayasu disease with high serum levels of IgA at onset. IgA remained elevated with persistence of disease activity, and normalized only when the patient was treated with an anti-TNF agent (infliximab), which also induced a clinical response in the vasculitis.
IgA levels, together with other inflammatory parameters, may be considered a biomarker of disease activity.
LEARNING POINTS
- This case report highlights the need to increase the number of humoral markers used to assess disease course in Takayasu arteritis (TA).
- IgA may be considered a biomarker of TA disease activity.
- Serum IgA levels may be helpful to identify TA patients not responding to traditional therapy.
KEYWORDS
Vasculitis, Takayasu arteritis, IgA, biomarkers, infliximab
INTRODUCTION
Takayasu arteritis (TA) is a rare inflammatory granulomatous vasculitis of the large vessels and mainly affects Japanese and Southeast Asian women in the second and third decades of life [1]. Inflammatory infiltrate affects the full thickness of the vessel wall, with remodelling and myofibroblast proliferation, followed by progressive lumen stenosis and occlusion [1]. Clinical manifestations are heterogeneous and depend on the involved anatomical region [1].
The pathogenesis of TA remains unknown, but environmental factors and genetic background contribute to its development [1, 2]. Innate and adaptative immune responses and proinflammatory circulating cytokines such as TNF-alfa, IL-6 and IL-8 have a pathogenetic role [2]. IL-6 plays a crucial role in tissue fibrosis genesis and its serum levels correlate with disease activity [2]. Other cytokines (IL-12, IL-18 and IFN-gamma) lead to granuloma formation [1, 2].
Patients with active disease have a higher erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) levels compared with patients with stable/inactive disease. Circulating IL-6 and IL-18 levels in patients with active disease tend to be higher than in those with stable/inactive disease or in healthy controls [3].
According to therapeutic guidelines, first-line therapy consists of high-dose (1 mg/kg daily) corticosteroids [1–3]. Other therapeutic strategies include disease-modifying anti-rheumatic drugs such as methotrexate, leflunomide and mycophenolate mofetil, or more aggressive immunosuppressive drugs such as cyclophosphamide. Biological drugs, such as anti-IL6 or anti-TNF-alfa agents, also appear to be effective in treating TA [1, 3, 4]
CASE DESCRIPTION
A 22-year-old woman from Bangladesh was hospitalized about 2 years ago for anaemia and scapulo-humeral pain radiating to the left arm and ipsilateral carotid region with strong carotidynia. She had headache, radial pulse loss and different arterial blood pressure readings between the upper arms.
Laboratory findings showed high inflammation indexes (ESR 120 mm/h, CRP >15 mg/dl, ferritinemia >800 µg/l) and hypergammaglobulinemia (serum protein immunofixation did not find monoclonal/oligoclonal components). At clinical onset, serum levels of IgA were elevated (1154 mg/dl, normal values 70–300 mg/dl), but those of the other Ig subclasses were not. During follow-up, IgA remained high, with fluctuation. Lymphocyte immunophenotyping showed increased CD3/4/8 T cell populations and increased CD3/HLA DR+, a marker of lymphocyte activation.
Computed tomography (CT) with contrast detected a filiform course of the left subclavian artery (maximum diameter 3 mm) and stenosis of the right renal artery. Positron emission tomography (PET) showed ‘intense metabolic activity on the carotid axis, extending to the right subclavian artery, the aortic arch and its root’, with a pattern of vascular metabolic pathology.
The patient was treated with high doses of prednisone (1 mg/kg/day) but developed Cushing’s disease with systemic arterial hypertension. Antimetabolites such as methotrexate and mycophenolate mofetil were poorly tolerated by the patient. Complete regression of the disease, of the radiological picture and of clinical features was never achieved.
Consequently, after 21 months, the patient was treated with anti-TNF monoclonal antibody therapy (infliximab), with one infusion every 6 weeks, and prednisone was gradually tapered to 0.3 mg/kg/day. IgA serum levels dropped to 260 mg/dl, with complete normalization of CRP (<0.5 mg/dl) and ESR (4 mm/h) (Fig. 1). The patient continued therapy with an antiplatelet agent (acetylsalicylic acid 100 mg every day).
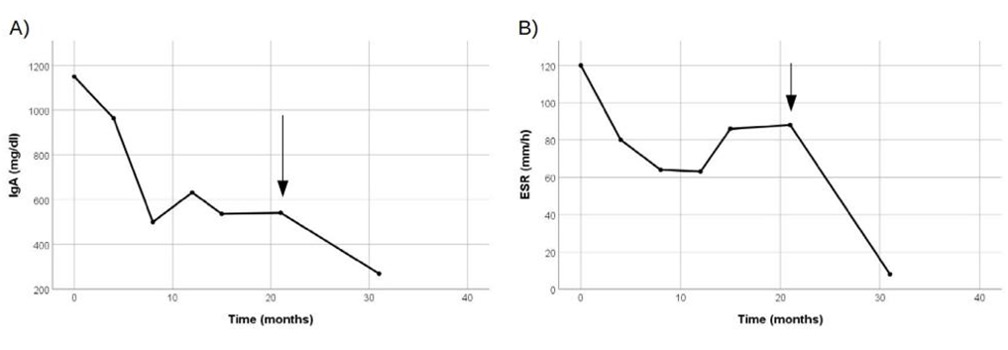
Figure 1 (click to enlarge)
Figure 1.IgA serum levels (panel A) and erythrocyte sedimentation rate (panel B). Arrows denote infliximab (300 mg) injection
DISCUSSION
IgA is the second most common Ig isotype in serum, where it is detected as a monomer. It is the most abundant Ig in mucosal tissue, where it is predominantly found as a dimeric protein. Dimeric IgA neutralizes pathogens, so it plays a crucial role in mucosal immunity, preventing the adhesion of viruses and bacteria to the mucous membranes [5]. Some pathogens can produce IgA proteases which degrade these mucosal antibodies, acting as virulence factors [6, 7]. Patients affected by selective IgA deficiency, which most commonly results in antibody immunodeficiency, have recurrent pulmonary infections caused by extracellular capsulated bacteria, and their small intestine can be colonized by Giardia lamblia, leading to bloating, cramping, excessive flatus and watery diarrhoea [8]. FcαRI, also known as CD89, is the main binding subset receptor for monomeric IgA and by the Fab region transduces inhibitory signals on antigen-presenting cells. It is expressed on neutrophils, monocytes, eosinophils and Kupffer cells [9]. IgA is a poor activator of complement compared with IgM and IgG and can block complement activation by binding other antibodies [9].
Two subclasses of IgA have been isolated: IgA1 and IgA2. In serum, monomeric IgA interacts with serum proteins as alfa-1antitripsine and could have a role for innate immune blocking neutrophils chemoattracting and inhibiting the pro-inflammatory response in monocytes. It can also bind albumin and fibronectin and thereby inhibit leukocyte elastase and neutrophil chemotaxis, playing an anti-inflammatory role. Serum IgA downregulates phagocytic leucocyte ability and pro-inflammatory cytokine release by peripheral blood mononuclear cells [7, 9].
Some authors have described cases of IgA-mediated nephropathy in TA and noted its incidence is higher in TA cases, but our patient did not have renal involvement [10, 11].
Microscopic polyangiitis (MPA) is a type of anti-neutrophil cytoplasmatic autoantibody-associated vasculitis. A retrospective study by Wang et al. focused on the correlation between IgA and readmission in MPA patients. The authors found that the risk of readmission was increased in patients with serum IgA levels >217.5 mg/dl [12].
Wei et al. performed a retrospective study to explore predictive factors associated with a poor TA prognosis and found that patients treated with cyclophosphamide had higher IgA levels but a lower rate of elevated CRP than non-responders [13].
CONCLUSION
The role of IgA is not limited to antibacterial and antiviral functions in mucosal immunity. Serum IgA can reduce cytokines and leucocyte, monocyte and neutrophil chemotaxis and send inhibitory signals to antigen-presenting cells. It could have an anti-inflammatory role or be a protective mechanism against tissue damage. Serum IgA levels may be helpful to identify patients not responding to traditional therapy.
Since the disease is rare, retrospective or prospective multicentre studies are required to determine if IgA could be a disease activity biomarker. It would be interesting to further investigate IgA function in Takayasu vasculitis pathophysiology.