ABSTRACT
Caroli disease is a rare congenital pathology caused by mutation of the PKHD1 gene (polycystic kidney and hepatic disease 1), also responsible for autosomal recessive polycystic kidney disease. Characterized by segmental and multifocal dilatation of the large intrahepatic bile ducts, classic disease involves only malformation of the biliary tract. The association with congenital hepatic fibrosis is called Caroli syndrome. We describe the case of an 84-year-old man with Caroli syndrome diagnosed in 1997 by liver biopsy. The CT scan revealed massive hepatomegaly, extending to the pelvic region, and almost total replacement of the parenchyma by numerous cystic formations, no evidence of bile duct dilatation, and no ascites or splenomegaly suggestive of portal hypertension. The atypical clinical presentation, with no reported complications, resembles that of a space-occupying lesion with an indolent course, previously misdiagnosed as metastatic neoplasm.
LEARNING POINTS
- We describe a case of advanced and rare Caroli syndrome with an atypical clinical presentation of a space-occupying lesion with slow progression.
- The atypical presentation could be misdiagnosed as metastatic neoplasm.
KEYWORDS
Caroli disease, polycystic kidney disease, hepatomegaly, intrahepatic bile ducts
CASE DESCRIPTION
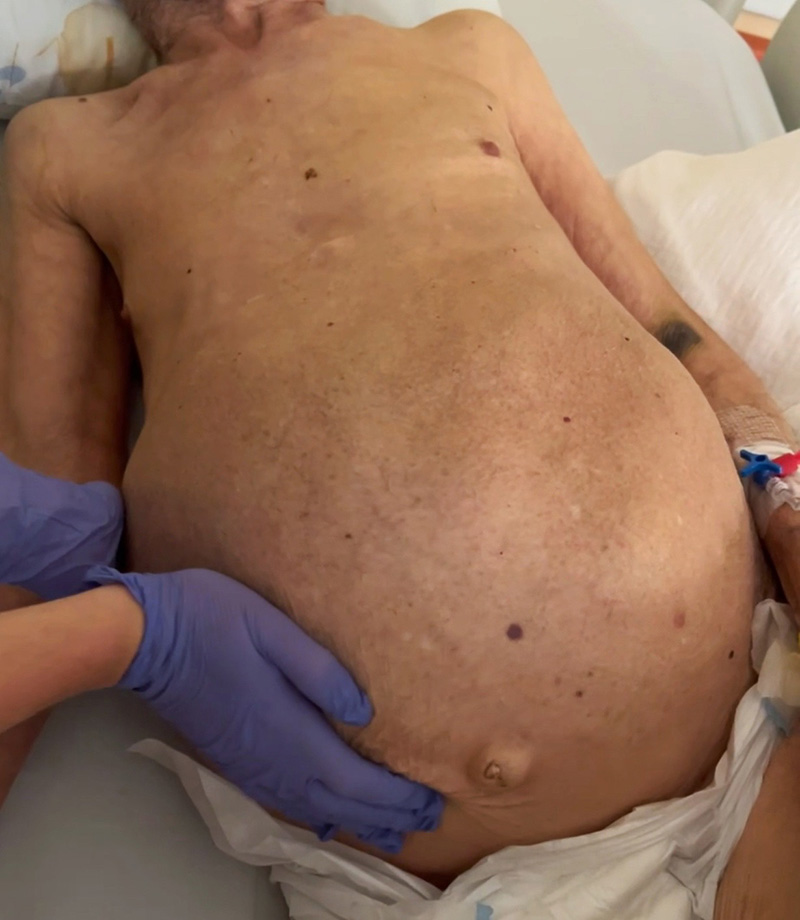
An 84-year-old man with dementia, prostate cancer and Caroli syndrome diagnosed in 1997 by liver biopsy, was admitted to the Emergency Department due to food aspiration. Upon admission, the patient presented with hypoxaemic respiratory failure and abdominal distention. On physical examination, there was evidence of massive hepatomegaly, with the hepatic border palpable at the level of the right iliac fossa, nodular contours, and firm consistency (Fig. 1). The patient was admitted to the Internal Medicine Service with a diagnosis of aspiration pneumonia. During the initial days of hospitalization, he continued to present symptoms caused by constipation that were unresponsive to medical therapy.
(click to enlarge)
Figure 1. Physical examination. Note the marked abdominal distension. Upon palpation, it was possible to detect massive hepatomegaly with the hepatic border at the level of the right iliac fossa, nodular contours, and firm consistency
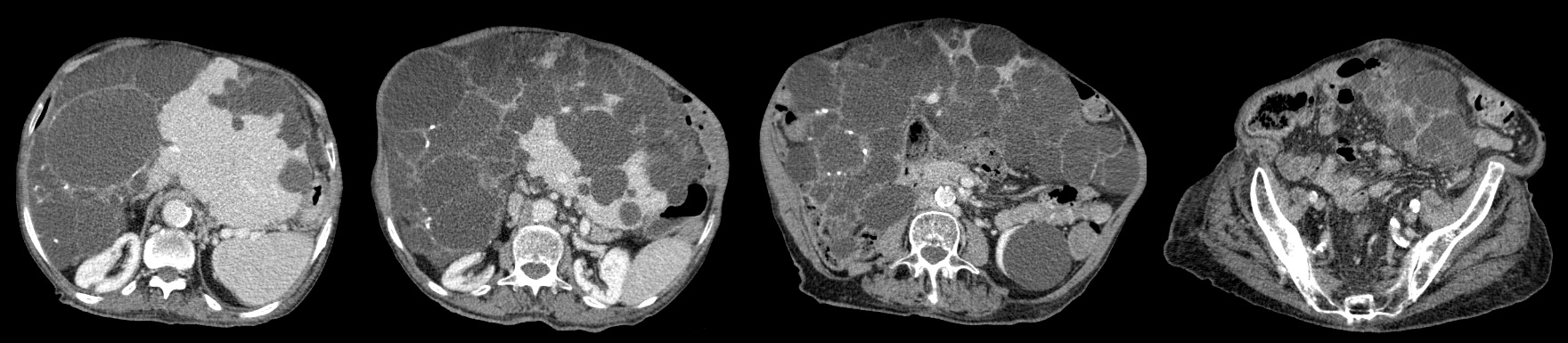
Due to his previous history of cancer, hepatomegaly and constipation, an abdominal and pelvic computed tomography (CT) scan was requested, to exclude intestinal occlusion and metastasis. The scan revealed massive hepatomegaly, extending to the pelvic region, and almost total replacement of the parenchyma by numerous cystic formations, without evidence of biliary tract dilation and without ascites or splenomegaly suggestive of portal hypertension (Fig. 2). Simple cortical renal cysts were also identified. There was no evidence of metastatic disease. Laboratory results demonstrated a pattern of cholestasis without an increase in bilirubin or transaminases, and normal kidney function.
(click to enlarge)
Figure 2. Abdominal and pelvic CT scan with contrast. Note the massive hepatomegaly characterized by almost total replacement of the hepatic parenchyma by cysts, with extension to the pelvic region and the presence of cortical renal cysts, without evidence of dilatation of the biliary tract
Mutation of the polycystic kidney and hepatic disease 1 (PKHD1) gene is responsible for both autosomal recessive polycystic kidney disease (ARPKD) and Caroli disease (chromosome 6p12, expressed primarily in the kidney and with lower levels in the liver and pancreas). The hepatic abnormalities occur later in life as there is relatively less expression of the affected fibrocystin gene in cholangiocytes compared with renal tubular cells[1,2].
On the other hand, polycystic liver disease most often occurs in patients with autosomal dominant polycystic kidney disease (ADPKD). This disorder is predominantly caused by mutations in one of two genes: PKD1 (which encodes polycystin-1) on chromosome 16 and PKD2 (which encodes polycystin-2) on chromosome 4. A less common disorder, autosomal dominant polycystic liver disease (PCLD), is distinct from polycystic kidney disease since it is not associated with kidney involvement[3].
Caroli disease is characterized by segmental and multifocal dilation of the large intrahepatic bile ducts. Classic disease involves only malformation of the biliary tract. However, an association with congenital hepatic fibrosis is more frequent and is called Caroli syndrome[4]. The diagnosis can be made by imaging or liver biopsy. Complications arising from Caroli syndrome include cholangitis, sepsis, choledocholithiasis, hepatic abscess, cholangiocarcinoma and portal hypertension[5]. Treatment is supportive, but if cholangitis coexists, antibiotic therapy or stenting through endoscopic retrograde cholangiopancreatography (ERCP) may be required[6]. In patients with extensive liver fibrosis, hepatectomy or liver transplantation may be considered. Many patients die within 5–10 years of developing cholangitis. Caroli syndrome may progress to cholangiocarcinoma[7]. The occurrence of hepatobiliary malignant transformation explained by chronic inflammation of the biliary tree, has been reported in 7–14% of patients. Death is related to liver failure or complications of portal hypertension[5]. Liver transplantation is the best treatment for this disease, and has a good prognosis, unless recurrent cholangitis and renal failure develop. In 2007, Millwala et al. conducted a study to determine the outcomes of liver transplantation in unselected patients with Caroli disease. Of the 78,124 patients who received a liver transplant in the USA between 1987 and 2006, 104 had Caroli disease; 96 of these underwent liver transplantation alone, and 8 underwent combined liver/kidney transplantation. The overall 1-year, 3-year and 5-year graft (79.9%, 72.4% and 72.4%) and patient (86.3%, 78.4% and 77%) survival rates were excellent for patients after liver transplantation. For combined liver/kidney transplantation (n=8), the 1-year patient survival and graft survival rates were 100%[8].
In the present case, the possibility of polycystic liver disease, associated with autosomal dominant polycystic kidney disease, had previously been excluded as the patient did not present with advanced kidney disease (normal kidney function) or a familial history suggestive of a dominant autosomal entity. Therefore, this is advanced and rare Caroli syndrome, and although there was exuberant replacement of the hepatic parenchyma by fibrosis and cystic formations, there was no portal hypertension, significant dilatation of the biliary tract, or a history of previous episodes of cholangitis. Therefore, the patient never had an indication for treatment, namely liver transplantation, or associated kidney dysfunction. Thus, with an atypical clinical presentation, the condition presented as a space-occupying lesion, with slow progression, in a geriatric patient, that could be misdiagnosed as metastatic neoplasm.