ABSTRACT
We report a case of a 19-year-old young male presenting with thyrotoxicosis with inappropriately elevated TSH. Magnetic resonance imaging revealed a pituitary adenoma (8.2 x 9.7 mm), TRH stimulation test showed abnormal blunted TSH response, and serum glycoprotein hormone alpha-sub-unit was elevated. He had no family history of thyroid disease and TRβ genetic testing excluded resistance to thyroid hormone action. The diagnosis of thyrotropin-secreting pituitary adenoma (TSHoma) was presumed and long-acting somatostatin analogue was promptly initiated. After two months of octreotide treatment, serum TSH and FT3 returned to within normal ranges. Tumour resection by transsphenoidal surgery was performed and, ten days after surgery, clinical hypothyroidism was achieved, despite detectable TSH levels (TSH 1.02 µU/ml[RR 0.27-4.2]). Although the patient remained euthyroid for the following three years, there was a gradual biochemical elevation in the levels of TSH, FT4, and FT3 over time, reaching serum values above the normal limit in the third year after surgery. Imaging did not show neoplasm recurrence at this point. After two years, the patient presented with clinical manifestations of re-onset thyrotoxicosis, with MRI revealing a T2 hypersignal oval area compatible with a pituitary adenoma. Adenectomy was performed. Histopathological and immunohistochemical analyses revealed a pituitary adenoma with transcription factor PIT1 expression and positivity for TSH and PRL. TSHoma treatment may not be always effective in the first therapeutic approach and recurrences are a possibility, making follow-up essential. The present case highlights the heterogeneity of post-treatment cure criteria and their limitations.
LEARNING POINTS
- Thyrotropin-secreting pituitary adenomas are rare benign tumours. Proper diagnosis can be challenging, requiring TSH autonomous production and differentiation from resistance to thyroid hormone action (RTH).
- Undetectable TSH levels one week after surgery and/or positive T3 suppression test or no response to TRH stimulation test seem to be the criteria with the best prognostic value post-treatment.
- Close clinical, biochemical and imaging follow-up is crucial to detect TSHoma recurrence..
KEYWORDS
Pituitary adenoma, thyrotoxicosis, hyperthyroidism, thyrotropin, neurosurgery
CASE DESCRIPTION
A 19-year-old young male with depressive disorder, sustained mood changes and involuntary weight loss was referred to our center in 2016 due to thyrotoxicosis with inappropriately elevated TSH (TSH 6.83 µU/ml [Reference Range (RR) 0.27-4.2], FT3 7.79 pg/ml [RR 2.0-4.4], FT4 1.76 ng/dL [RR 0.93-1.7]).
Further investigation showed heterogeneous micronodular thyroid echostructure and high radioactive iodine homogeneous uptake on thyroid scintigraphy, with negative thyroid autoantibodies. Magnetic resonance imaging (MRI) revealed a pituitary tumour (8.2 x 9.7 mm), TRH stimulation test showed abnormal TSH blunted response and serum glycoprotein hormone alpha-sub-unit (α-GSU) (1,17 mUI/ml [RR 0.00-0.80]) and α-GSU/TSH molar ratio (α-GSU/TSH>1) were elevated, suggesting a thyrotropin-secreting pituitary adenoma. There was no family history of thyroid disease and TRβ genetic testing excluded resistance to thyroid hormone action (RTH).
TSHoma diagnosis was made and long-acting somatostatin analogue was promptly initiated. After two months of octreotide treatment, serum TSH and FT3 returned to within normal ranges (TSH 4.27 µU/ml [RR 0.27-4.2], FT3 4.54 pg/ml [RR 2.0-4.4], FT4 1.25 ng/dl [RR 0.93-1.7]). The patient underwent tumour resection by transsphenoidal surgery (TSS) and, ten days after surgery, clinical hypothyroidism was achieved, despite detectable TSH serum levels (TSH 1.02 µU/ml [RR 0.27-4.2], FT3 1.69 pg/ml [RR 2.0-4.4], FT4 0.50 ng/dl [RR 0.93-1.7]). At this point, the patient started levothyroxine supplementation.
Histopathological analyses showed adeno- and neurohypophysis fragments with normal characteristics and no neoplasm identified.
Although the patient remained euthyroid for the following three years, there was a gradual biochemical elevation in the levels of TSH, FT4, and FT3 over time, reaching serum values above the normal limit in the third year after surgery (TSH 4.62 µU/ml [RR 0.27-4.2], FT3 4.42 pg/ml [RR 2.0-4.4]), instantly raising the suspicion of TSHoma recurrence. Imaging did not show neoplasm recurrence at this point.
Two years later, in 2021, during the COVID-19 pandemic, the patient exhibited significant mood changes and panic attacks with re-onset of thyrotoxicosis with inappropriately elevated TSH (TSH 5.03 µU/ml [RR 0.27-4.2], FT3 8.35 pg/ml [RR 2.0-4.4], FT4 1.89 ng/dl [RR 0.93-1.7]). MRI now revealed a T2 hypersignal oval area of approximately 8 mm, on the anterior and right lateral aspect of the adenohypophysis, allied with a slight increase in volume/height of the pituitary gland on that side and slight deviation of the pituitary stalk, compatible with an adenoma (Fig. 1).
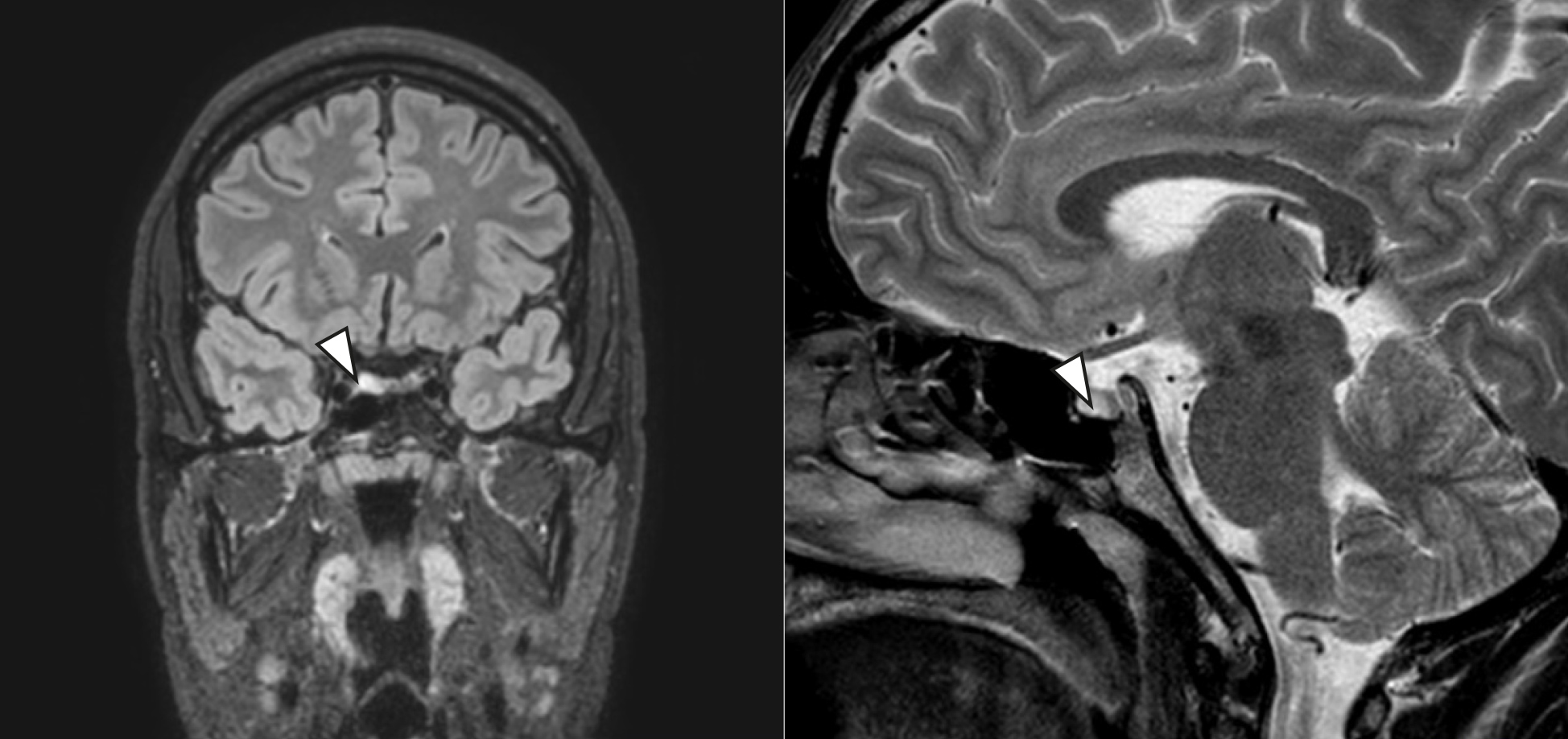
(click to enlarge)
Figure 1. T2-weighted MRI - oval area of T2 hypersignal (closed arrow), with approximately 8 mm, on the anterior and right lateral aspect of the adenohypophysis, allied with a slight increase in volume/height of the pituitary gland on that side and a slight deviation of the pituitary stalk, compatible with an adenoma.
Transsphenoidal adenectomy was performed for a second time and hypothyroidism with minimal serum TSH levels (TSH 0.09 µU/ml, FT3 1.41 pg/ml, FT4 0.34 ng/dl) was accomplished postsurgery. Histopathological and immunohistochemical analyses finally exposed a pituitary adenoma with transcription factor PIT1 expression and positivity for TSH and PRL. Genetic testing for AIP mutation was negative.
Surgical cure of this rare nosological entity was assumed and the patient is currently clinically euthyroid. The patient is maintaining annual clinical and analytical examination.
DISCUSSION
Thyrotropin-secreting pituitary adenomas are rare benign entities, accounting for 0.5-3% of all pituitary adenomas, demanding appropriate investigation[1,2]. RTH, an autosomal dominant disorder, is the main differential diagnosis, manifested with high FT4 and FT3 levels in the presence of non-supressed TSH, which contrasts with the biochemical pattern founded in primary hyperthyroidism[2]. Our case exemplifies the diagnostic course of “inappropriate TSH secretion syndrome” and how challenging its treatment can be.
Pathological molecular mechanisms leading to TSHoma formation are still unknown. Familial cases as part of multiple endocrine neoplasia type 1 syndrome (MEN-1) and familiar isolated pituitary adenoma family (FIPA) related to AIP mutations are described in the literature[3]. In our patient, genetic test for AIP mutations was negative and genetic test for MEN-1 syndrome was deferred, despite his young age, considering the absence of other clinical entities integrated in MEN-1 syndrome.
The majority of patients present with a pituitary adenoma in MRI, but some ectopic localizations in nasopharyngeal and suprasellar regions have been also reported[4]. Dynamic testing through TRH stimulation test (200 µg i.v.) or T3 suppression test (80-100 µg/day for 8-10 days) should be performed for definitive diagnosis of TSHoma. Blunted TSH responses to TRH stimulation occur in more than 80% of TSHomas and complete suppression of TSH from feedback administration of T3, indicating autonomic secretion, has never been reported[2].
The European Thyroid Association guidelines[3] recommend transsphenoidal or subfrontal adenectomy as the first line of treatment to normalize thyroid function. Despite a high rate of complete adenoma removal in patients with microadenomas (<10 mm), surgical outcomes remain unsatisfactory in the presence of marked fibrosis and in the case of invasive macroadenomas, cavernous sinus invasion being a predictive factor for compromised extension of surgical tumour resection[1]. Even so, surgical strategy can restore euthyroidism in more than 80% of patients, with a possible consequence of partial/complete hypopituitarism[4]. Long-term alternatives to surgical treatment are pituitary radiotherapy and somatostatin analogs. Somatostatin receptors are expressed in almost all TSHomas[3]. It is expected that chronic administration of long-acting somatostatin analogs for at least 2 months decreases FT4 and FT3 levels in TSHoma, with no response in patients with RTH. Chronic administration was associated with the attainment of euthyroidism in 90% of patients and significant tumour shrinkage. Additionally, some cases of TSHoma cure with medical therapy are described[3].
Post-treatment cure criteria have not been distinctly established and show highly heterogeneity[3]. Remission from hyperthyroidism manifestations, normalization of free thyroid hormone, α-GSU and TSH levels, undetectable TSH levels one week after surgery and/or a positive T3 suppression test and no response to TRH have been proposed as cure criteria, although with different limitations[1]. As shown by our clinical case, remission of hyperthyroid symptoms and thyroid function normalization may be transient and not synonymous of complete destruction of tumours cells. Neuroradiological revaluation performed soon after surgery is frequently difficult to interpret. Still, undetectable TSH levels one week after surgery and/or positive T3 suppression test and no response to TRH stimulation test seem to be the criteria with the best prognostic value[1,5]. In our patient, despite achieving clinical hypothyroidism, detectable TSH and progressive resurgence of TSHoma in the four years post-surgery made it difficult to consider TSH normalization as cure criteria.
TSHoma treatment is not always effective in the first therapeutic approach, highlighting the importance of close clinical, biochemical and imaging follow-up. The present case highlights the possibility of long-term future recurrence. In this case, TSHoma reoccurred four years after the surgical intervention. This was detected by the medical team, allowing the establishment of a new therapeutic intervention.