ABSTRACT
We report a case of a 64-year-old man who, 44 days after starting treatment with prasugrel, presented with severe thrombocytopenia, anaemia, renal failure, and severe ADAMTS13 activity deficiency, along with a high titer of autoantibodies to this protease.
LEARNING POINTS
- Drug-induced TTP is a rare condition and difficult to diagnose.
- Decreased activity of ADAMTS13, unusual in drug-induced TTP, was present in this case.
KEYWORDS
Thrombotic microangiopathy; thrombotic thrombocytopenic purpura; prasugrel.
INTRODUCTION
Thrombotic thrombocytopenic purpura (TTP) is defined as a severe deficiency of ADAMTS13, but the diagnosis of TTP relies initially on clinical judgment, since ADAMTS13 measures are often not available for several days, and different methodologies may yield different results. ADAMTS13 can be completely deficient, genetically impaired or inhibited by autoantibody. It is now observed that all acquired TTP, including those cases of acquired TTP that are drug induced, are associated with inhibiting antibodies to ADAMTS13. TTP is unique among the primary thrombotic microangiopathy (TMA) syndromes, in which renal function abnormalities occur but renal failure is rare. TTP typically has more systemic manifestations of organ injury than the other primary TMA syndromes. In TTP, abnormalities of the central nervous system, heart, pancreas, thyroid glands, adrenal glands, intestinal mucosa and other tissues may occur.
CASE REPORT
A 64-year-old male patient with a medical history of hypertension, type 2 diabetes mellitus (DM), and dyslipidemia was admitted to hospital due to a ST-elevated coronary syndrome in 2013. He underwent fibrinolysis with tenecteplase (metylase) without complications, followed by placement of a TAXUS drug-eluting stent, placed into the anterior descending coronary artery. He was started on prasugrel 10 mg daily and discharged from the hospital on day 12 after admission.
The patient was readmitted to hospital 36 days later due to nausea, vomiting, abdominal pain, and purpuric lesions. Upon arrival to the emergency department, he was hypotensive and oliguric. An abdominal computed tomography (CT) was immediately performed and was positive for acute appendicitis. The patient underwent surgery, and the surgeons noted haemoperitoneum and acute appendicitis, with the presence of a mass compatible with abscess. After surgery, laboratory tests revealed progressive anemia, severe thrombocytopenia, acute renal failure (serum creatinine 3.64 mg/dL) (Figs. 1, 2 and 3), and increases in creatine kinase (CK), lactose dehydrogenase (LDH), procalcitonin, C-reactive protein (CPR), and indirect bilirubin. Laboratory indicators of disseminated intravascular coagulation were negative.
Upon arrival, prasugrel was discontinued, and he was started on low molecular weight heparin considering the previous medical history. Piperacillin was initiated in addition to volume expanders and blood products. Forty-eight hours after surgery, his chemical panel showed a decrease of inflammatory markers, CPR and procalcitonin and improvement of haemoglobin, platelets and renal function.
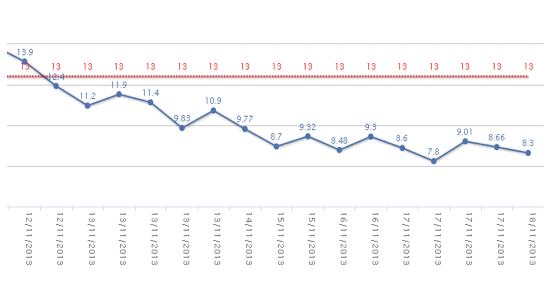
Figure 1 (click to enlarge)
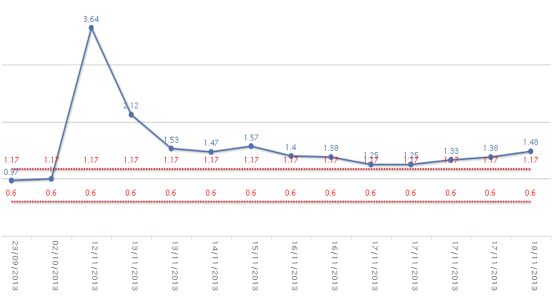
Figure 2 (click to enlarge)
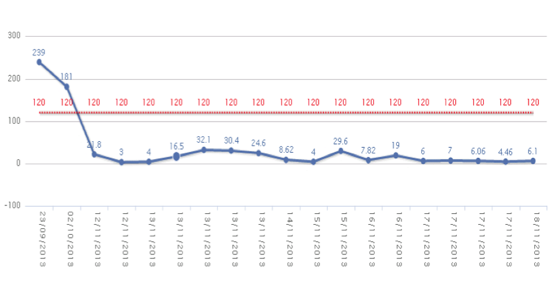
Figure 3 (click to enlarge)
Figure 2. Serum creatinine (mg/dL) curve behavior. Plasmapheresis was initiated 4 days after admission.
Figure 3. Platelets curve behavior. Plasmapheresis was initiated 4 days after admission.
On day 3 after surgery, the patient reported headaches preceding mental confusion and stupor. No abnormalities were found on the CT scan or on the electroencephalogram, and the patient recovered spontaneously. Forty-eight hours later, the patient became hemodynamically unstable and presented difficulty speaking and transient numbness of the right arm. Laboratory tests were ordered showing anemia, thrombocytopenia, and 6% schistocytes on the peripheral blood smear. Direct Coombs test was negative. PT, PTT and fibrinogen were within the normal range. Thrombocytopenic purpura was diagnosed, in view of the entire clinical picture, and plasmapheresis was initiated. Despite daily plasmapheresis, the patient’s neurological condition progressed to coma and death.
ADAMTS13 levels were at 0.3% and inhibitory antibodies were at high titers. No bacteremia was identified. The autopsy revealed thrombi in the microcirculation, mainly affecting the adrenal glands, spleen, heart, bone marrow, and bowels (Fig. 4). Subendothelial deposits were not found.
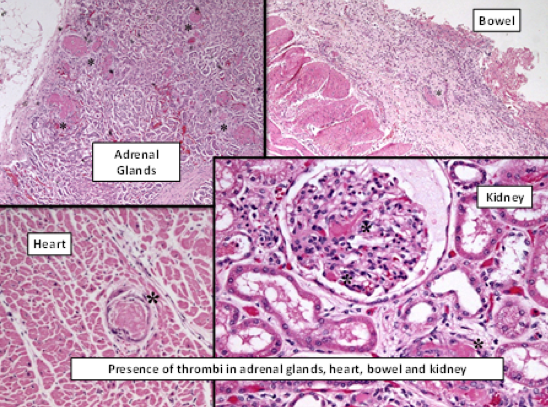
Figure 4 (click to enlarge)
DISCUSSION
TMA induced by drugs is an acquired condition, secondary to antibody production. Tissue damage may also result from the direct formation of thrombi in small arterioles and capillaries. Organ infarction may occur because of thrombosis in small vessels, and kidneys seem to be particularly susceptible[1]. While in some publications the term “drug-induced TTP” is widely used, recent studies have preferred to use this term to define cases of TTP in which there is an ADAMTS13 activity deficiency[1]. Drug-induced TTP is uncommon and difficult to diagnose, and the mechanisms leading to it are not clear in most cases[2].
The Oklahoma TTP-HUS Registry, conducted between 1998 and 2014 and including 487 patients, only described 5% of cases of drug induced TTP[3]. A study carried out in the US between 1991 and 2011 stated that clopidogrel was most commonly associated with TTP (197 cases), followed by ticlopidine (97 cases) and prasugrel 14 cases[4]. Interestingly, TTP is mainly described as appearing early after drug initiation. In our case, clinical and laboratory findings consistent with TTP appeared 44 days after prasugrel was initiated.
The mechanisms of drug-induced TTP are considered to be either direct tissue toxicity or through immunologically-mediated syndromes[1]. Some drugs are believed to produce a direct endothelial injury and this effect is directly associated with the dosage used[2,3], while immunologically-mediated syndromes are considered to be idiosyncratic. It is unusual for cases of drug-induced TTP to show a severely decreased activity of ADAMTS13 (activity <10%), yet a relatively normal or slightly decreased activity in the absence of inhibitory antibodies would help in its diagnosis. Thienopyridines are the only drugs pathophysiologically related to the presence of neutralizing antibodies against ADAMTS13 and present a good response to plasmapheresis[4].
Even though 14 cases of prasugrel-related TTP have been reported by the FDA, there has been no evidence until now of the biological significance of ADAMTS13 activity, as shown in our patient. Ticlopidine-related TTP appears from 2 to 12 weeks after the drug is initiated and presents with severe thrombocytopenia and acute renal failure, similar to our case. There are no confirmative laboratory tests for drug-induced TTP, so the diagnosis must be made only by considering the clinical history and the specific previous drug exposure of the patient[2].
The identification of drug-dependent antibodies may help diagnose this entity, but their absence may not exclude the diagnosis[3].
Clinically, patients with TTP and severe ADAMTS13 deficiency rarely present with important neurological findings and renal impairment. In addition, coronary revascularization and surgery have also been related to the development of TTP. Plasmapheresis can help not only to remove auto-antibodies, cytokines, and multimers of von Willebrand factor, but also to replace ADAMTS13[5].
CONCLUSIONS
Prasugrel appears to have caused TTP in this reported case, since there were no other laboratory or histological findings consistent with sepsis or disseminated intravascular coagulation.
Unfortunately, we were not able to study the complement levels or carry out genetic studies.