ABSTRACT
Creutzfeldt-Jakob disease (CJD) is a rare, incurable and fatal condition that can only be confirmed through neuropathological investigation, such as brain biopsy or post-mortem study. However, a probable diagnosis can be made using clinical criteria. CJD manifests as rapidly progressive dementia with myoclonus and to a lesser extent visual impairment and cerebellar and pyramidal/extrapyramidal signs. We report the case of a previously independent adult male that met all the clinical criteria. Taken together, the investigation results suggested probable CJD.
LEARNING POINTS
- Creutzfeldt-Jakob disease (CJD) is a rare cause of dementia.
- The rapidly progressive neurological signs and symptoms suggest the diagnosis.
- Mortality rates are very high even with surgical treatment in these complex patients.
KEYWORDS
Creutzfeldt-Jakob disease; rapid progressive dementia
INTRODUCTION
Creutzfeldt-Jakob disease (CJD) is a rare, incurable and inevitably fatal neurodegenerative condition, and is the most frequent human prion disease[1].
There are four recognized forms of CJD: familial CJD (fCJD), iatrogenic CJD (iCJD), variant forms of CJD (vCJD) and sporadic CJD (sCJD), with the latter being responsible for the majority of reported cases[2].
Despite the rarity of the disorder, which has an annual incidence rate of 1–1.5 people per million worldwide annually[3], authorities have tried to define diagnostic criteria for CJD. The Center for Disease Control and Prevention (CDC) and Zerr classification criteria for CJD describe definite CJD as disease diagnosed by neuropathological techniques and/or immunocytochemically, Western blot-confirmed protease-resistant PrP, and the presence of scrapie-associated fibrils.
Probable CJD can be diagnosed when all routine diagnostic examinations exclude an alternative diagnosis and there is rapidly progressive dementia, two out of four clinical features (myoclonus, visual or cerebellar signs, pyramidal/extrapyramidal signs and akinetic mutism) and a positive result in at least one of the following laboratory tests: 14-3-3 found in the cerebrospinal fluid (CSF) of patients with a disease duration of less than 2 years; a typical EEG (periodic sharp wave complexes); or magnetic resonance imaging (MRI) high signal abnormalities in caudate nucleus and/or putamen on diffusion-weighted imaging (DWI) or fluid attenuated inversion recovery (FLAIR).
Possible CJD is defined when patients present with the same clinical features but all of the above examinations are negative, illness duration is less than 2 years, and there are no routine investigations indicating an alternative diagnosis[4].
CASE REPORT
A 57-year-old, previously independent white man presented to the emergency department (ED) with a 2-month history of left-sided headache, dizziness and progressive visual disturbances. He also mentioned recent memory loss and gait imbalance without preferential fall. These progressive neurological changes had motivated several previous visits to the ED, but no alterations in neurological and ophthalmological examinations had been found. However, at the last visit, he presented with right homonymous hemianopsia, horizontal bilateral inexhaustible nystagmus and broad-based petit-pas gait with progressive altered mental status and hetero-aggressiveness. This required markedly increased sedation, subsequent intubation and ventilation of the patient and transfer to our intensive care unit (ICU). None of the three ED head CT scans or other laboratory examinations showed any alterations.
The patient worked in a slaughterhouse, where he was in direct contact with cattle, and ingested approximately 12 g of alcohol daily. His other past history was not considered relevant for the anamnesis.
Initially, the authors considered the following differential diagnoses: infectious encephalitis, auto-immune encephalitis, metabolic encephalitis secondary to alcohol consumption, sarcoidosis and malignancy, which were all excluded after the diagnostic investigations described below. CJD was the primary differential diagnosis because of the epidemiological context.
Laboratory results showed CSF with elevated glucose of 82 mg/dl (normal range 40–70 mg/dl), normal protein, chloride and lactate dehydrogenase levels, and less than 3 leukocytes/mm3, without red cells. CSF was positive for 14-3-3 protein and cultures were negative. Initially the patient had a modest elevation of C-reactive protein coincident with a nosocomial methicillin susceptible Staphylococcus aureus pneumonia that normalized after treatment with flucloxacillin.
The autoimmunity study revealed negative titres of anti-double-stranded deoxyribonucleic acid (anti-ds-DNA), antinuclear (ANA), antineutrophil cytoplasmic (ANCA), anti-neuronal, anti-myelin, anti-ganglioside, anti-recoverin, anti-SOX1 and all other autoantibodies included in the dementia differential diagnosis.
MRI images showed areas of hyperintensity on T2 bilaterally in the putamen (with greater extent on the left side) (Fig. 1) and in the subcortical parietal lobe with diffusion restriction (Fig. 2). Electroencephalographic study showed slow and disorganized brain electrical activity, with clips of periodic or generalized pseudo-periodic activity indicating diffuse encephalopathy (Fig. 3).
The patient was discharged from the ICU to a neurology ward and died in the hospital 4 months after the initial symptoms. The family did not give consent for an autopsy.
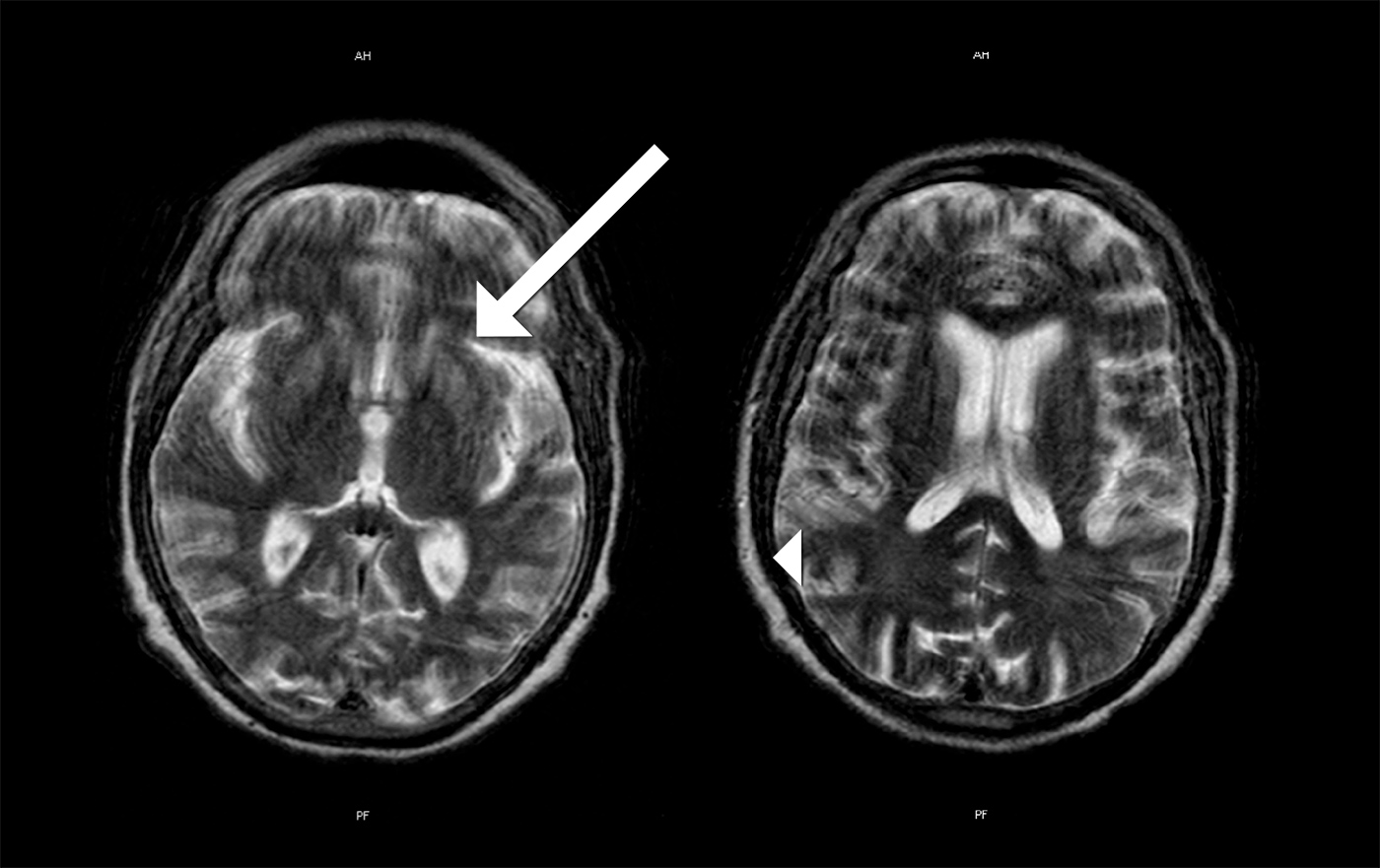
Figure 1 (click to enlarge)
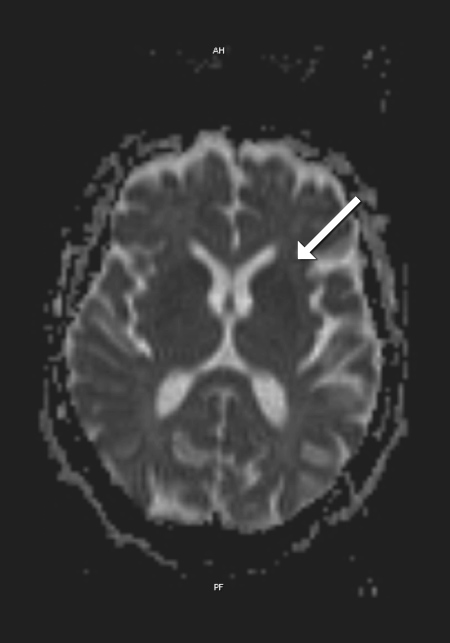
Figure 2 (click to enlarge)
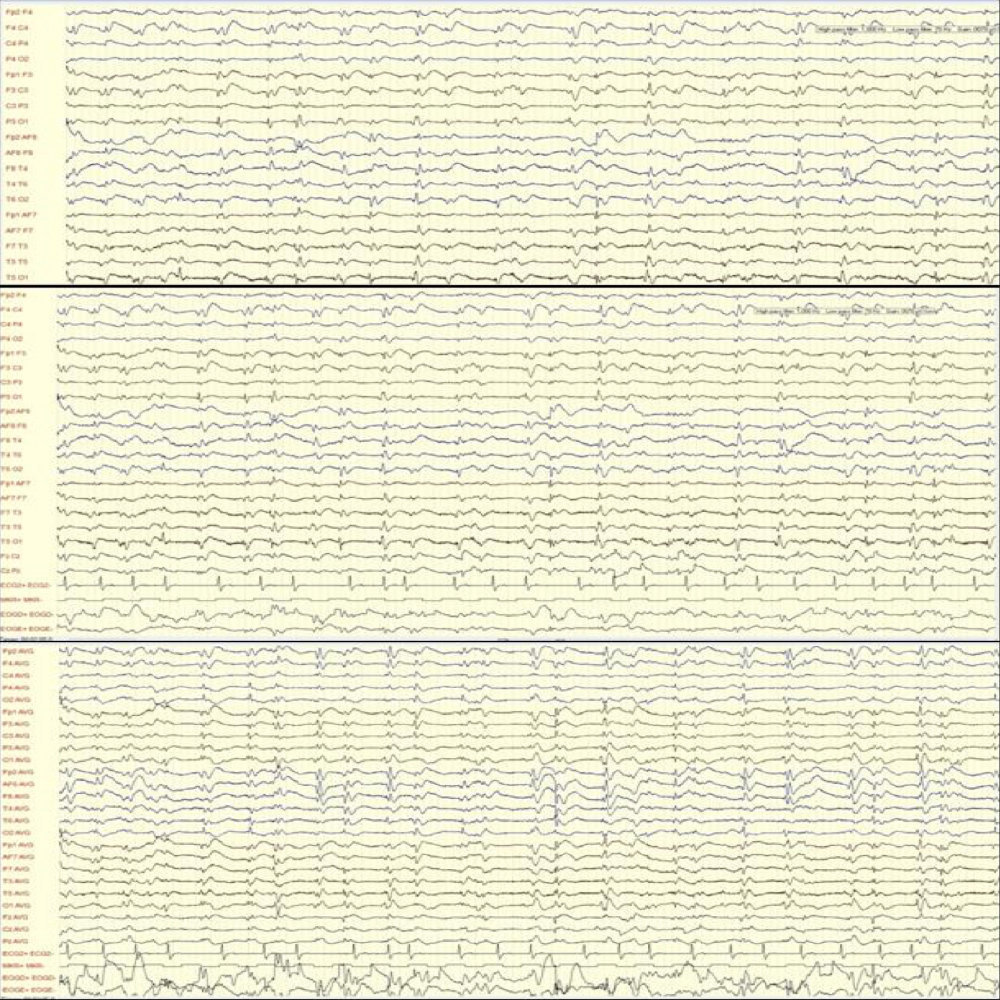
Figure 3 (click to enlarge)
Figure 1. MRI images showing hyperintensity on T2 bilaterally in the putamen with greater extent on the left side (arrow) and in the subcortical parietal lobe (arrow head)
Figure 2. MRI images of diffusion restriction in the putamen (arrow)
Figure 3. Electroencephalographic images showing slow and disorganized brain electrical activity, with clips of periodic or generalized pseudo-periodic activity
DISCUSSION
According to the CDC criteria, our patient had probable CJD. As the family refused an autopsy, a definite diagnosis of CJD was impossible. However, since there is no screening test for CJD, a definite diagnosis is of no use, but a diagnosis of probable CJD is sufficient to raise epidemiological awareness. Despite our patient's rapid clinical course, the family did not seem concerned with the possibility of another CJD case.
Although CJD is a rare condition, sporadic cases of the disease are still seen with variant Creutzfeldt-Jakob disease (vCJD) occurring less often. Because of the bovine spongiform encephalopathy epidemic in the 1990's, the extended incubation period of approximately 10–20 years[5] and the patient's type of employment, vCJD is a probable diagnosis[2].
The clinical course progressed to death in just 4 months from the initial symptoms. The literature[1,2,5] refers a mean age of 57–62 years at disease onset and a median survival of 6 months with death occurring usually within 1 year of symptom onset. vCJD differs from sCJD in that it occurs at a younger age and has slower disease progression[5], which reduces the probability of vCJD in our patient.
In conclusion, CJD is a rare disease that should be considered in adult patients with rapidly progressive dementia.