ABSTRACT
Porphyrias are a group of disorders caused by enzymatic defects in the biosynthesis of haem. Congenital erythropoietic porphyria (CEP) or Günther's disease is an extremely rare autosomal recessive disorder. Clinical manifestations include onset in infancy of blistering of sun-exposed areas, atrophic scars, mutilated fingers, and bright red fluorescence of the urine and teeth without neurological involvement. The only available prophylactic treatment for CEP is total avoidance of sunlight. A high degree of suspicion is required for the diagnosis. Early diagnosis is very important to prevent subsequent damage.
LEARNING POINTS
- A rare case of congenital erythropoietic porphyria (CEP) without evidence of haemolysis is described.
- A high degree of suspicion is required for diagnosing CEP.
- Early diagnosis prevents further damage.
KEYWORDS
Congenital Erythropoietic Porphyria, Porphyrins, UROS
INTRODUCTION
A case of congenital erythropoietic porphyria (CEP) with a classic clinical presentation but without haemolysis is described.
CASE REPORT
A 12-year-old boy born of a third-degree consanguineous marriage to a nomad family from Rajasthan, India was admitted to the ophthalmology department of our hospital with left eye scleromalacia requiring scleral grafting. He had undergone scleral grafting in the right eye for a similar indication the previous year. He was referred to the Department of Internal Medicine for evaluation of photosensitive skin rashes since early childhood.
There was a history of recurrent blisters over exposed parts of the body such as the face, neck, forearms, hands and feet since 6 months of age. Blisters would develop and heal within few days leaving scarred and disfigured skin (Fig. 1). Blisters forming over the tips of fingers and ear lobules would be followed by mutilation (Fig. 2). His parents also noticed him passing reddish brown urine since infancy.
There was no history of drug intake, acute abdominal pain, acute neurological attacks or blood transfusion. His birth and developmental history were normal. However, there was a tradition of consanguineous marriages in the family, with his elder brother and his father's younger brother also experiencing a similar illness since early childhood.
On examination, the patient had normal intelligence and mental function. His anthropometric parameters were normal for his age and sex. His systemic examination was unremarkable.
Skin examination revealed blisters limited to sun-exposed areas of the body. Some of the blisters were at the healing stage and his face was badly scarred. The skin of hairy scalp and covered parts of body was normal. The face, neck, hands and feet showed extensive scarring, multiple vesicles 1–3 mm in diameter and hyperpigmentation with foci of hypopigmentation. There was hypertrichosis on the face and forearms. Atrophic scars resulted in mutilation and disfigurement with contractures of the fingers of both hands (Fig. 3). Nails were dystrophic. Eye examination showed left nasal scleromalacia and an old right temporal scleral graft. There was no corneal scarring. Reddish brown staining of the teeth was present. The urine had reddish brown hue on standing (Fig. 4).
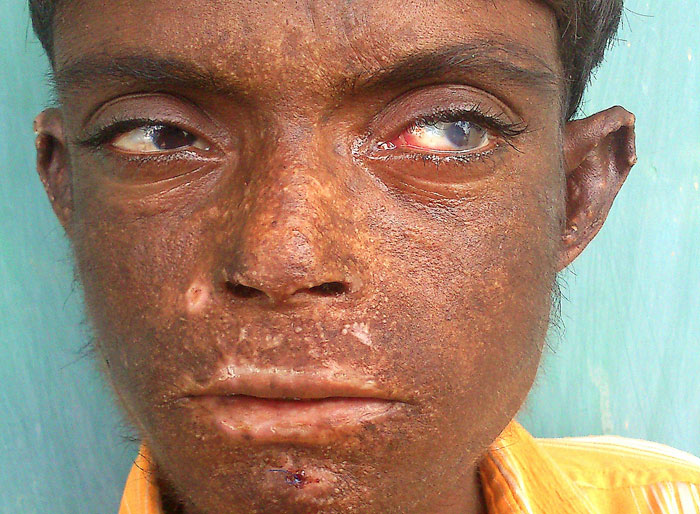
Figure 1 (click to enlarge)
Figure 1. Vesicles, scarring, pigmentation, hypertrichosis on the face and scleromalacia of the left eye
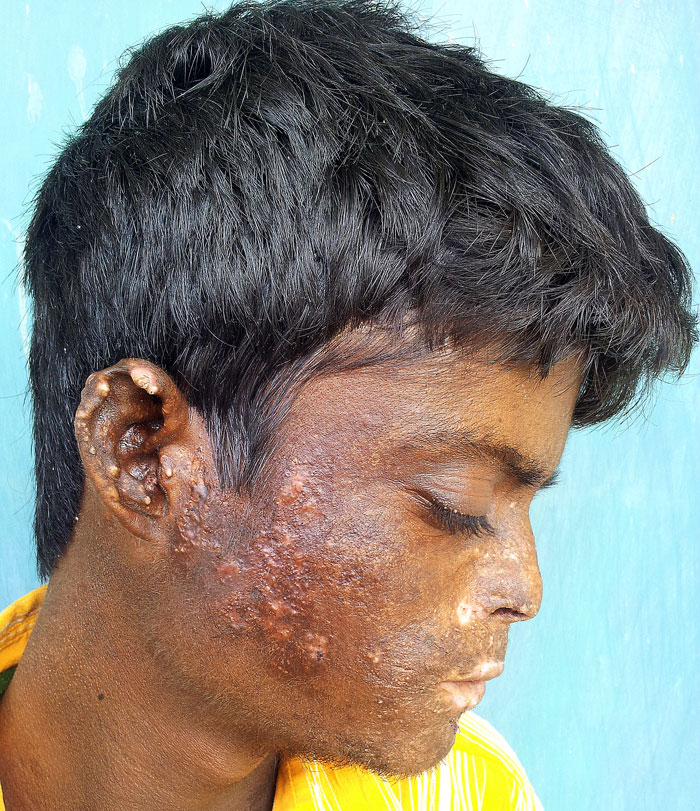
Figure 2 (click to enlarge)
Figure 2. Vesicles, scarring, pigmentation, hypertrichosis of the face and mutilation of the ear lobule
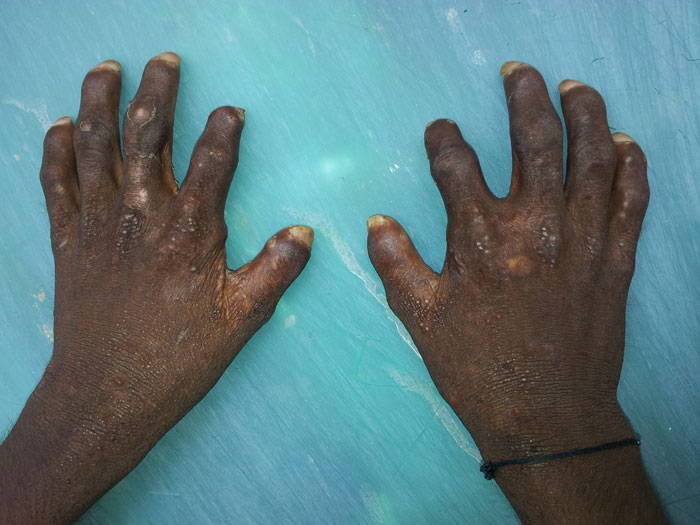
Figure 3 (click to enlarge)
Figure 3. Vesicles, scarring of the hands, mutilating deformities of the fingers and dystrophic nails.

Figure 4 (click to enlarge)
Figure 4. Urine turning reddish pink on standing
Bright red fluorescence of the urine and teeth was noted under a Wood's lamp. The patient's haemoglobin was 13 g/dl with a normal peripheral smear and blood indices. Reticulocyte count was 1% of circulating erythrocytes with a negative Coombs test. Routine examination and microscopy of the urine were normal. Liver and renal function tests were normal and serology for HIV was negative. Ultrasonography did not reveal any hepatosplenomegaly or gallstones.
We made a provisional diagnosis of CEP on the basis of infancy onset of blistering of sun-exposed areas, atrophic scars, mutilated fingers, and bright red fluorescence of the urine and teeth without neurological involvement.
Column chromatography showed a urinary total porphyrin level of 1550 nmol/mmol of creatinine (normal <35 nmol/mmol). Urine did not show increased levels of delta aminolevulinic acid or porphobilinogen. X-ray of the fingers showed acro-osteolysis (Fig. 5). The erythrocyte porphyrin level and URO synthase deficiency could not be assessed or genetic tests carried out due to lack of resources and financial constraints.
A final diagnosis of CEP was made. We advised strict avoidance of sunlight and prescribed topical sunscreen lotion to prevent further damage. The child was treated symptomatically but subsequently lost to follow-up after the first visit.
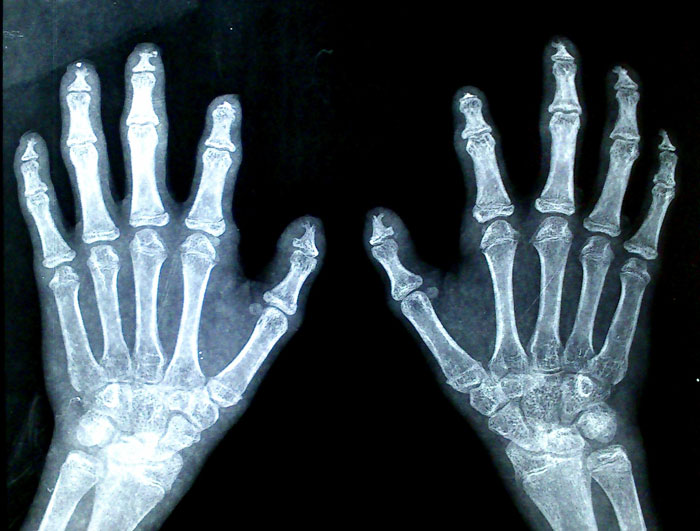
Figure 5 (click to enlarge)
Figure 5. X-ray showing acro-osteolysis of the fingers
DISCUSSION
Porphyrias are a group of disorders caused by enzymatic defects in the biosynthesis of heme. Clinically, porphyrias are classified as cutaneous photosensitivity (e.g. CEP), neuropsychiatric (e.g. acute intermittent porphyria) and both cutaneous and acute disease (e.g. variegate porphyria). CEP or Günther's disease is an extremely rare autosomal recessive disorder with fewer than 200 cases reported in the literature until recently. A mutation on chromosome 10q26.2 causes markedly deficient activity of the enzyme uroporphyrinogen III (URO) synthase in erythrocyte precursor cells in bone marrow, resulting in massive accumulation of uroporphrin-I and coproporphyrin-I in bone marrow, red blood cells (RBCs), skin, bones, teeth, urine and faeces. Porphyrins absorb light photons of around 408 nm wavelength and cause cell damage to surrounding structures when they transfer energy as they return to their normal positions[1].
Cutaneous photosensitivity to sunlight and other sources of ultraviolet light usually begins in early infancy and is manifested by increased friability and blistering of the epidermis on the hands and face and other sun-exposed areas. Red-brown staining of nappies due to markedly increased urinary porphyrins may be the first indication of the disease. The skin may be thickened, with areas of hypo- and hyperpigmentation. Recurrent vesicles and secondary infection lead to cutaneous scarring, deformities and mutilation of the fingers, nails, eyelids, nose and ears. Hypertrichosis of the face and extremities may often lead to what is sometimes called werewolf syndrome. Scleromalacia and corneal ulcers lead to blindness. Erythrodontia (red discoloration of the teeth under ultraviolet light) when present is virtually pathognomonic of CEP[2].
Haemolysis is due to increased fragility of RBCs caused by accumulation of porphyrins and is characterised by anisocytosis, reticulocytosis, absence of haptoglobin, increased unconjugated bilirubin and faecal urobilinogen. Anaemia can be so severe that some patients are dependent on transfusions.
Epidermolysis bullosa and drug-induced pseudoporphyria can mimic CEP due to the presence of extensive photosensitivity and mutilation. However, in both of these conditions, porphyrin levels in the urine, plasma and faeces are normal. Histopathological examination of bullae in CEP shows caterpillar bodies which are composed of eosinophilic linear basement membrane material in the roof of sub-epidermal blisters. Urinary porphyrin excretion is markedly increased (up to 50–100 mg/day) and consists mostly of uroporphyrin and coproporphyrin. Urinary aminolevulinic acid and porphobilinogen excretion is not increased. Faecal porphyrins are markedly increased due to increased coproporphyrin I. Circulating erythrocytes contain large amounts of uroporphyrin I and coproporphyrin I. A definitive diagnosis of CEP can be made by demonstration of markedly deficient URO synthase activity[3].
The only available prophylactic measure for CEP is total avoidance of sunlight. Sun-protective clothing, topical sunscreen lotions, use of CFLs instead of incandescent bulbs and oral photoprotectants such as beta-carotene 120–180 mg/day are advisable to decrease oxidative damage. Other treatments include intravenous haematin therapy to suppress haemoglobin production by negative feedback, oral activated charcoal to prevent reabsorption, and hydroxyurea to reduce bone marrow porphyrin synthesis. Despite the limited treatment options, life expectancy is 40–60 years[4].
Specific mutations in the UROS gene can be identified with genetic testing. Allogenic bone marrow transplantation replaces erythroblasts in the medulla and reverses disease manifestations but is costly. Stem cell transplantation is currently the only known curative therapy and has great promise[5].
In our case there was infancy-onset cutaneous photosensitivity, infancy-onset blistering, and red-coloured urine and teeth together with cutaneous scarring. However, strikingly, there was no evidence of haemolysis. This unique presentation encouraged us to report such a rare case. A high degree of suspicion is required for the diagnosis of CEP and the early recognition is very important to prevent future damage.