ABSTRACT
Background: Some patients exhibit features of both autoimmune hepatitis (AIH) and primary sclerosing cholangitis (PSC). Similarly, patients with progressive familial intrahepatic cholestasis type 3 (PFIC3) may share histological features with PSC.
Case report: We report the case of a 22-year-old man who, since he was 5 years of age, has presented with pruritus, an approximately ninefold elevation of aminotransferases, and γ-glutamyl transferase levels ~10 times the upper limit. Initially he was diagnosed with an overlap syndrome of small duct PSC plus AIH. However, fluctuations in liver enzymes were observed over the following years. Analysis of the ABCB4 gene indicated the diagnosis of PFIC3, revealing a mutation not previously reported.
Conclusion: With this case report we aim to describe a new mutation, raise awareness of this rare pathology and highlight the importance of genetic testing of the ABCB4 gene in patients with autoimmune liver disease (mainly small duct PSC) with incomplete response to immunosuppressive treatment.
LEARNING POINTS
- Autoimmune liver diseases have a wide spectrum of manifestations.
- Cholangiopathies such as ABCB4 deficiency have histological features quite similar to those seen in small duct primary sclerosing cholangitis.
- The new mutation of the ABCB4 gene described in this article is compatible with the diagnosis of progressive familial intrahepatic cholestasis type 3, which is probably less rare than usually thought.
KEYWORDS
ABCB4; autoimmune hepatitis; primary sclerosing cholangitis; progressive familial intrahepatic cholestasis
INTRODUCTION
Autoimmune liver diseases have a wide spectrum of manifestations. Some patients present with features of both autoimmune hepatitis (AIH) and cholestatic diseases: primary sclerosing cholangitis (PSC) or primary biliary cholangitis (PBC). AIH is a complex disease of the hepatic parenchyma characterized by increased activity of serum aminotransferases, hypergammaglobulinaemia, circulating autoantibodies and interface hepatitis on liver histology[1,2]. PSC is a cholestatic liver disease of unknown aetiology characterized by an inflammatory and fibrotic process affecting intra- and extra-hepatic bile ducts[3]. The diagnosis of PSC is assumed in patients with elevated serum markers of cholestasis, serum alkaline phosphatase (AP) or γ-glutamyl transferase (γ-GT), and typical bile duct changes revealed by magnetic resonance cholangiography (MRC) or endoscopic cholangiopancreatography (ERCP)[3,4]. Some patients have clinical, biochemical and histological features compatible with PSC, but a normal image study, and are classified as having small duct PSC[5].
The co-existence of features of AIH and features of PSC, commonly referred as 'overlap syndromes', has been described in children and adults[6,7]. Around 10% of patients with typical AIH have histological features of bile duct injury, thus making the diagnosis of small duct PSC unclear[8] Likewise, patients with other cholangiopathies such as ABCB4 deficiency have histological features quite similar to those seen with small duct PSC[9]. Progressive familial intrahepatic cholestasis type 3 (PFIC3), caused by a genetic defect on the ABCB4 gene, belongs to a heterogeneous group of autosomal recessive liver disorders, with childhood predominance, which causes cholestasis of hepatocellular origin[10].
CASE REPORT
We describe the case of a 22-year-old man admitted to our outpatient clinic when he was 11 years old. He had presented with pruritus without jaundice since he was 5 years of age and his past medical records revealed elevation of alanine (ALT) and aspartate (AST) aminotransferase up to nine times the upper limit (UL) and of γ-GT up to ten times the UL. His family history was unremarkable except for the presence of PBC in his paternal grandmother (Fig. 1).
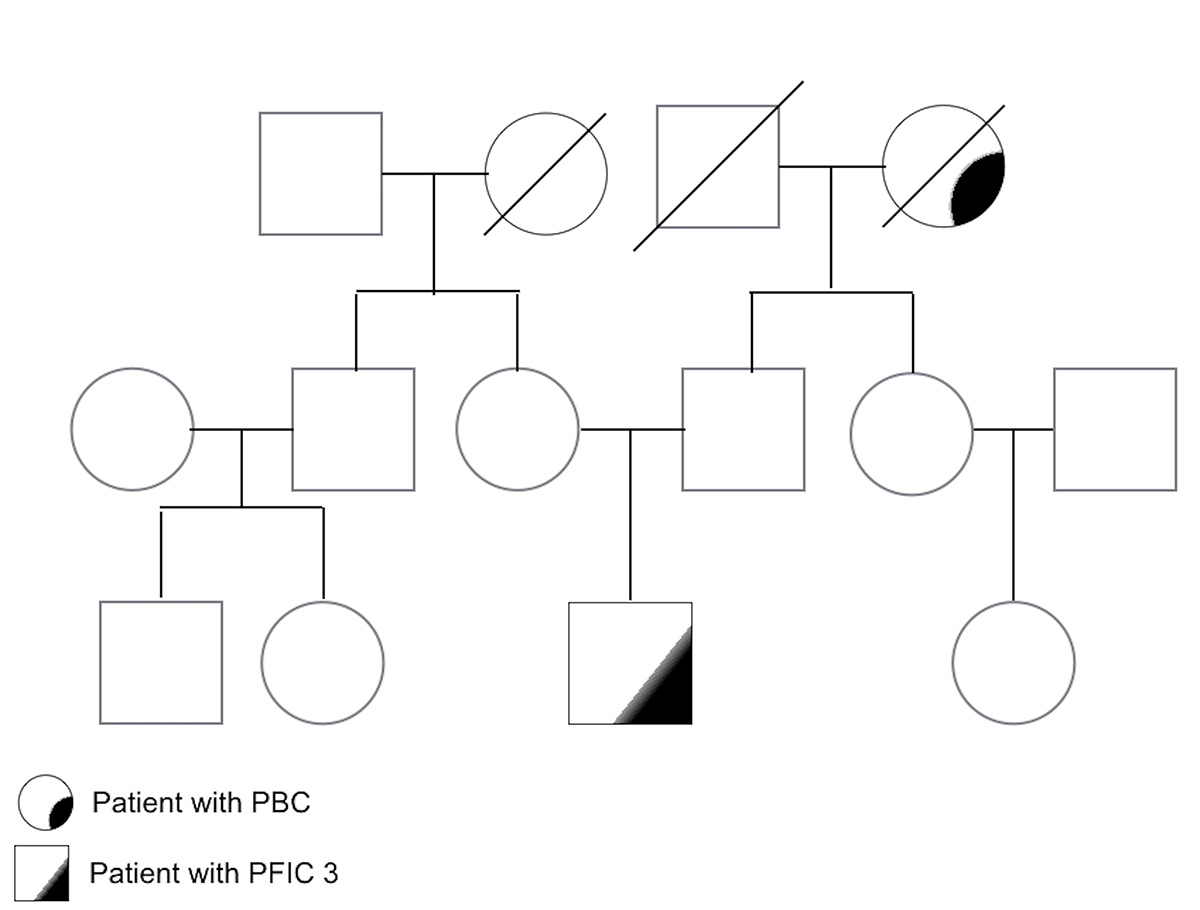
Figure 1 (click to enlarge)
Figure 1. Family genogram
On admission, blood tests revealed: total bilirubin 0.84 mg/dl, AST 96 IU/l, ALT 165 IU/l, γ-GT 219 IU/l and AP 577 IU/l. Ultrasound and MRC showed a normal sized and shaped liver, with normal intra- and extra-hepatic bile ducts. Immunological tests revealed: IgG 1440 mg/dl, antinuclear antibody 1/1280 with a nucleolar pattern and an anti-smooth-muscle antibody 1/160. Liver histology revealed preserved lobular architecture, portal space expansion due to polymorphic inflammatory infiltrate, a ductal lesion with lymphocytic invasion and occasional periductal fibrosis and ductular proliferation but no interface hepatitis. Nevertheless, a trial of immunosuppression was started with prednisolone and azathioprine without improvement of aminotransferase levels and persisting high levels of γ-GT and AP. The diagnosis of small duct PSC was proposed, azathioprine and prednisolone were withdrawn and the patient was treated with ursodeoxycholic acid, exhibiting fluctuating levels of cholestasis (Fig. 2).
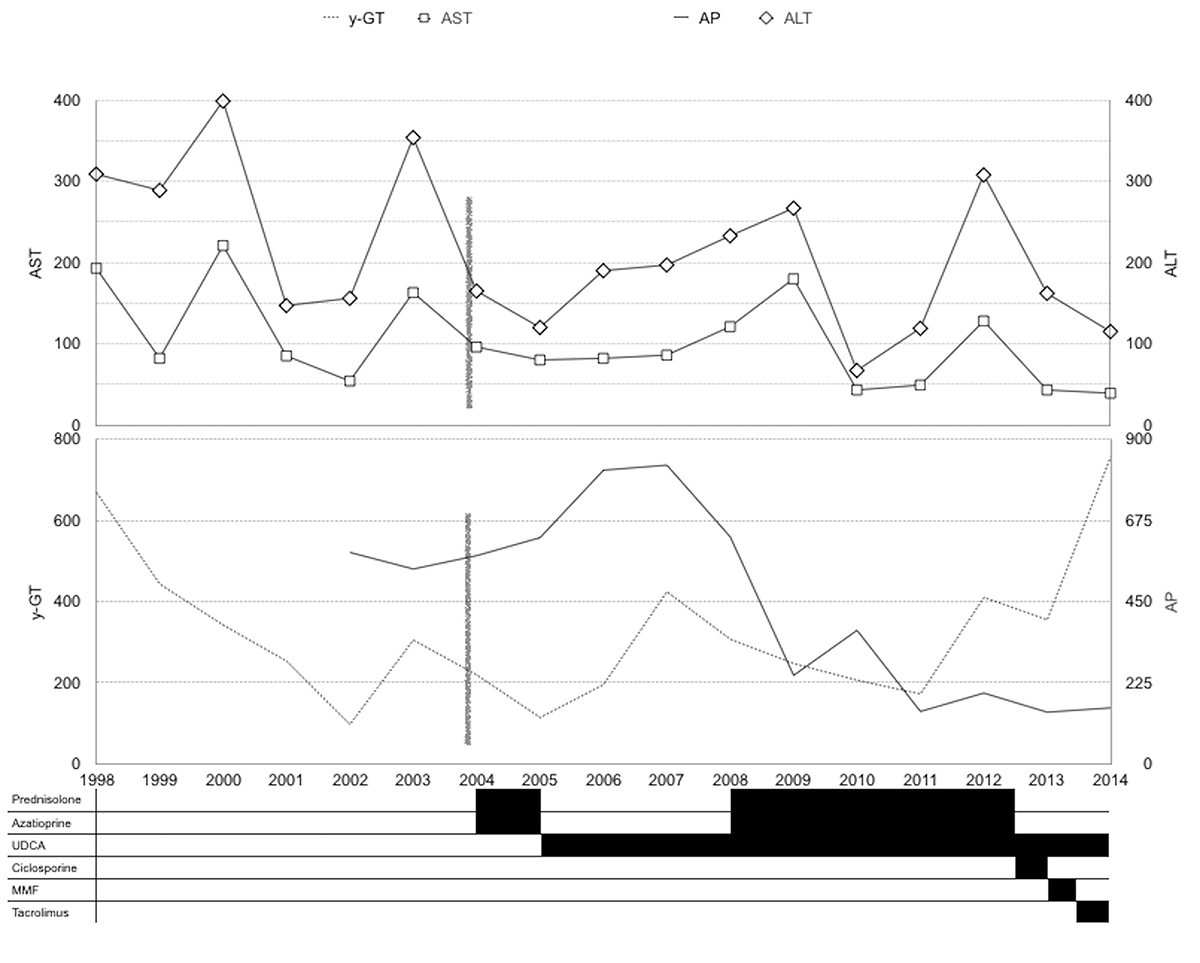
Figure 2 (click to enlarge)
Figure 2. Evolution of serum aminotransferases and cholestatic enzymes
Four years later he presented with an increase in AST and ALT levels and a similar immunological study. A second liver biopsy (Fig. 3) showed portal expansion with polymorphic inflammatory infiltrate with lymphocytic predominance and interface and intra-lobular necro-inflammatory activity. The diagnosis of an overlap syndrome (small duct PSC+AIH type 1) was made and prednisolone plus azathioprine were started with normalization of AST and ALT. Over the following years, we observed fluctuations in liver enzyme levels (Fig. 2), unrelated to the levels or type of immunosuppression.
We searched for mutations on the ABCB4 gene. Complete sequencing of the ABCB4 gene was performed using the sequence NM_000443.3. The patient had two mutated alleles in heterozygosity: c.874A>T and c.3680T>C. The genetic study of his parents showed the presence of heterozygous mutations in the ABCB4 gene: c.874A>T (p.Lys292*) in his father and c.3680T>C (p.Ile1227Thr) in his mother. The global analysis revealed that the biallelic distribution of the ABCB4 mutants in our patient is compatible with the diagnosis of PFIC3, which due to a variant not previously described.
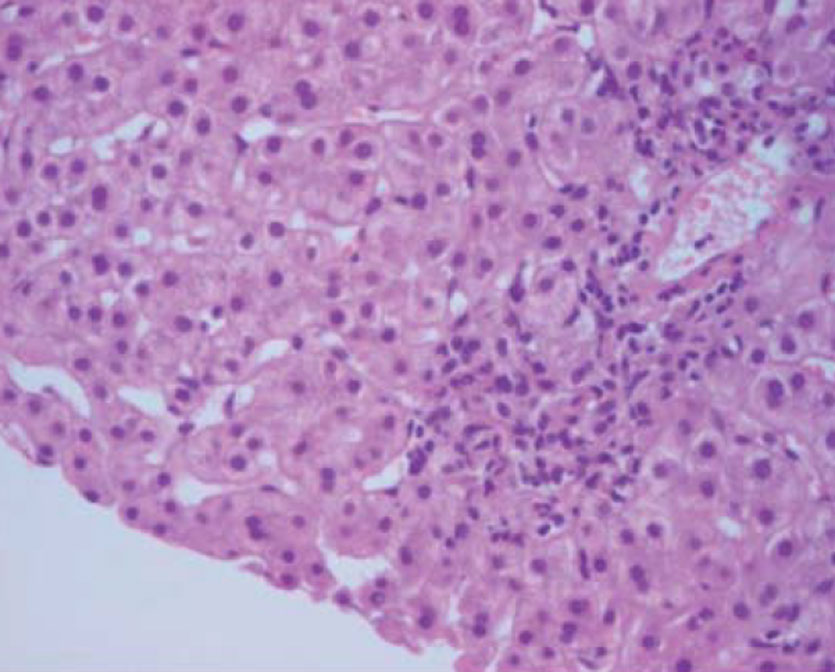
Figure 3 (click to enlarge)
Figure 3. Liver histology showing interface and intra-lobular necro-inflammatory activity
DISCUSSION
This case shows that some patients do not fit into a specific disease. Initially, this patient was considered to merely have small duct PSC due to the low levels of AST and ALT associated with the absence of interface hepatitis and no response to immunosuppression. Various reports have stated that some patients, mainly children, may present with cholangiopathies with features similar to those of AIH[11]. The diagnosis of small duct PSC is still being discussed, but a liver biopsy is mandatory for the diagnosis; however, liver histological findings of PSC are non-specific[4].
The pattern of AST and ALT noticed 4 years later in our patient caused our group to revaluate the first diagnosis. Based on the autoantibodies found and the presence of interface hepatitis and reinforced by more recent studies[6,7], we assumed that this patient had an 'overlap syndrome' and was presenting with features of PSC and AIH.
Maintenance of the same enzymatic pattern, unrelated to the modalities or intensity of immunosuppression, led us to search for mutations on the ABCB4 gene and results were compatible with the diagnosis of PFIC3. PFIC3 patients complain of recurrent pruritus and/or jaundice with chronic high levels of serum γ-GT, typically 10 times the UL[12].
The cholestatic pattern observed in our patient, which for a long time was ascribed to small duct PSC, is the result of PFIC3. Our patient had an allele with a previously described mutation[13] and a new genetic variant: c.3680T>C (p.Ile1227Thr), which has not been previously reported, in the allele transmitted by his mother. This allele is reported in the ExAC database to have a global frequency of 0.00082%, with bioinformatics analysis indicating that is a deleterious mutation.
Our patient, who for a long time was considered to have a PSC-AIH overlap syndrome, had a rare cholestatic disease (PFIC3) and AIH type 1. The interesting association of PFIC3 and AIH type 1 has not been described in the literature. Nonetheless, the association PFIC3-AIH may be fortuitous and PFIC3 is probably less rare than usually thought. We report this case with the aim of raising awareness of this rare pathology and highlighting the importance of genetic testing of the ABCB4 gene in patients with autoimmune liver disease (mainly small duct PSC) with incomplete response to immunosuppressive treatment.