ABSTRACT
Antiphospholipid syndrome (APS) is an autoimmune disease which can be primary or secondary to other autoimmune conditions and is defined by the occurrence of arterial or venous thrombosis, or pregnancy morbidity associated with persistently positive antiphospholipid antibodies (aPLA). The kidney may be affected by thrombosis at any level of its vasculature. When small vessels are involved, this results in thrombotic microangiopathy (TMA), which can manifest as either acute vaso-occlusive or chronic vascular lesions in glomeruli, arterioles and interlobular arteries. We report the case of 26-year-old man, with a previous medical history suggestive of APS, who was found to have a small elevation in serum creatinine. A kidney biopsy was performed and revealed features of chronic TMA. Anticoagulation was begun and kidney function remained stable. However, one year later, upon suspension of anticoagulation, the patient developed acute kidney injury and a second kidney biopsy showed acute TMA. This case describes different manifestations of antiphospholipid syndrome nephropathy (APSN) and highlights the importance of anticoagulation for thrombosis prevention.
LEARNING POINTS
- Identification of the aetiological factors of tender erythematous nodules is challenging.
- Acute kidney injury due to acute thrombotic microangiopathy (TMA) may develop in a patient with chronic TMA, often after a precipitating event. In the case reported here, the most likely trigger was the suspension of anticoagulation after a surgical emergency.
- Long-term anticoagulation is the mainstay of treatment for both APS and APSN; discontinuing anticoagulation increases the risk of thrombosis and has to be carefully weighed against the risk of serious haemorrhage.
KEYWORDS
Antiphospholipid syndrome; antiphospholipid syndrome nephropathy; kidney biopsy; thrombotic microangiopathy
INTRODUCTION
Panniculitis is an inflammation of the subcutaneous adipose tissue under the skin epidermis, and usually presents with inflammatory nodules or plaques[1]. A wide variety of panniculitis subtypes exists, mainly in the geriatric population, including panniculitis related to infection, external insults, malignancy, inflammatory diseases and enzymatic destruction. This wide range of possibilities may delay the diagnosis, leading to prolonged hospitalization. Recognizing important diagnostic clues may pinpoint the correct disease, even before its onset.
CASE PRESENTATION
A 26-year-old Caucasian man was admitted to the orthopaedics department in May 2015 for a spinal fracture sustained in a road accident. He had a medical history of focal epilepsy diagnosed 10 years earlier, episodes of amaurosis fugax, and positive measurements in previous years for the three antiphospholipid antibodies (aPLA). Cerebral magnetic resonance imaging performed in 2013 suggested lacunar ischaemic foci and he was started on aspirin. The patient had also had a non-seminoma testicular tumour that was treated with radical inguinal orchiectomy and four cycles of bleomycin, etoposide and cisplatin which ended 4 months before admission. His ongoing medication included aspirin, zonisamide, oxcarbazepine and levetiracetam. He had no known kidney disease and denied urinary symptoms. At admission, his serum creatinine was 1.2 mg/dl. He had a tonic-clonic seizure in the emergency room, underwent three contrast-enhanced computed tomography (CT) scans during his first few days in hospital and was started on amoxicillin/clavulanic acid for a presumed respiratory infection. Ten days after admission, his creatinine had risen to 1.7 mg/dl and urine showed haematuria and a urine protein/creatinine ratio of 0.4 g/g. On ultrasound, the kidneys showed normal size, differentiation and vascular resistance and no signs of obstruction. An immunological study was negative except for the three aPLA. A kidney biopsy was performed (Fig. 1).
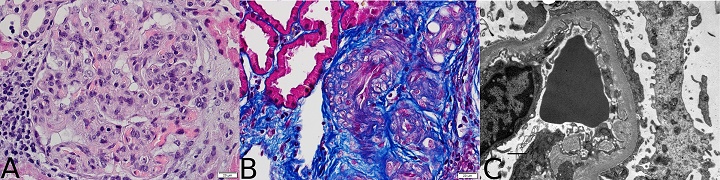
Figure 1 (click to enlarge)
Figure 1. (A) Glomerulus with membranoproliferative features: increased lobulation, intracapillary hypercellularity and thickening of the capillary walls (haematoxylin-eosin ×400); (B) arteriolar tortuosity with a narrow lumen (trichrome staining ×400); (C) expansion of lamina rara interna and duplication of the glomerular basement membrane (electron micrograph ×12,000)
The biopsy showed membranoproliferative features in one of 20 glomeruli, small artery tortuosity and severe intimal fibrosis. Immunofluorescence was negative, while electronic microscopy showed clear expansion of lamina rara interna. Taken together with the patient's history, these features were suggestive of chronic TMA in the context of APS. The patient started anticoagulation with warfarin, hydroxychloroquine and lisinopril, and during the following year maintained a stable serum creatinine of 1.2–1.4 mg/dl and had unremarkable urine examinations. One year later, upon suspension of anticoagulation after a physical attack resulting in spleen rupture, he developed an acute kidney injury and needed haemodialysis. He was submitted for urgent splenectomy and warfarin was stopped on the day of admission. During the first week in hospital, two contrast-enhanced CT scans were performed and vancomycin was empirically begun for raised inflammatory markers and fever, although all blood cultures were negative and the patient was always haemodynamically stable. Serum creatinine increased one week after admission and laboratory values also displayed anaemia, thrombocytopenia, and elevated lactate dehydrogenase with no other signs of haemolysis. Additionally, an echocardiogram showed aortic valve calcification and severe dysfunction. Kidney function continued to worsen during the following week with oliguria. The patient developed acute pulmonary oedema, was subsequently intubated and ventilated, and started renal replacement therapy. Because of the lack of recovery of kidney function after extubation, a new kidney biopsy was performed and revealed acute TMA with fibrin thrombi occluding the lumen of small arteries (Fig. 2). Anticoagulation was resumed with unfractionated heparin followed by warfarin; the patient recovered from dialysis after 3 months and now has a serum creatinine of 2.0 mg/dl.
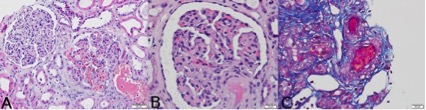
Figure 2 (click to enlarge)
Figure 2. (A) Membranoproliferative features, glomerular capillary congestion, haemorrhage and acute tubular injury (haematoxylin-eosin ×200); (B) afferent artery hyalinosis (haematoxylin-eosin ×400); (C) severe intimal fibrosis and thrombus in the arterial lumen (trichrome staining ×400)
DISCUSSION
APS is a systemic autoimmune disorder characterized by at least one clinical criterion (vascular thrombosis or pregnancy morbidity) and a positive aPLA test in measurements >12 weeks apart[1]. APS may be primary or secondary to another autoimmune disease. Frequent clinical manifestations include deep vein thrombosis, pulmonary embolism, transient ischaemic attacks/stroke, thrombocytopenia, livedo reticularis, cardiac valve thickenings/vegetations and epilepsy[2]. Our case fits primary APS: the patient had epilepsy, amaurosis fugax and valvular dysfunction, consistently positive aPLA and an immunological study that does not suggest any other autoimmune disorder.
Renal involvement may be due to renal large vessel thrombosis or small vessel vaso-occlusive nephropathy, a condition called antiphospholipid syndrome nephropathy (APSN). Presentation is unspecific and may include hypertension, proteinuria or increased serum creatinine. Very rarely, APS behaves aggressively with multiorgan failure, a form known as catastrophic APS. Our patient's first biopsy was unremarkable, featuring endothelial damage but no clear vascular occlusions. At that time, he did not have any laboratory signs of haemolysis. The second biopsy revealed acute TMA which caused acute kidney failure. Anticoagulation withdrawal was considered the major precipitating factor, but the surgery and presumed infection likely also contributed to thrombosis.
Treatment consists of lifelong efficient anticoagulation. The risk of bleeding has to be weighed against the benefits of anticoagulation. Our patient had postoperative conditions indicating early anticoagulation should be started; however, the thrombotic risk was undervalued. There are other more controversial therapies such as the use of hydroxychloroquine to reduce thrombotic risk, renin-angiotensin-aldosterone system antagonists to control proteinuria and hypertension, or the management of catastrophic APLS with additional corticosteroids, plasmapheresis, rituximab or eculizumab[3].