ABSTRACT
Senile systemic amyloidosis is caused by a non-mutated form of transthyretin with the heart being the major organ involved. This infiltrative cardiomyopathy usually presents as slowly progressive heart failure.
An 82-year-old female patient was admitted for newly diagnosed heart failure. A year later she presented with decompensated heart failure and syncope. Inpatient work-up showed persistently elevated troponin and N-terminal-pro BNP levels, rapid progression to severe left ventricular concentric hypertrophy, and sinus pauses on the Holter. Cardiac MRI revealed diffuse late gadolinium enhancement in the left ventricle. The demonstration of amyloid protein with the clinical findings and complementary investigations allowed for the diagnosis of senile systemic amyloidosis.
LEARNING POINTS
- Senile systemic amyloidosis is a rare disease with a common clinical presentation that is probably underdiagnosed in patients with heart failure with preserved systolic function.
- A new unexplained heart failure diagnosis, increased ventricular wall thickness and particularly low voltage on the ECG should raise the suspicion of cardiac amyloidosis.
- Histopathological evidence of amyloid deposition elsewhere and typical advanced imaging features can support the diagnosis, rendering endomyocardial biopsy no longer mandatory.
KEYWORDS
Cardiac amyloidosis, senile systemic amyloidosis, amyloidosis, heart failure with preserved ejection fraction
INTRODUCTION
Amyloidosis is a rare disease characterized by extracellular tissue deposition of protein fibrils with an aberrantly folded beta-pleated sheet configuration. Senile systemic amyloidosis (SSA) is a subtype of amyloidosis where the protein precursor is wild-type transthyretin (TTR), usually presenting as a restrictive cardiomyopathy.
The authors describe a case of cardiac amyloidosis in an elderly woman admitted for heart failure and syncope, an unusual aetiology for common symptoms at such an age.
CASE REPORT
An 82-year-old active female patient was admitted to the emergency department for asthenia, progressive fatigue with minor effort, and orthopnoea for the past several days. The patient had no chest pain, cough or fever. Additionally, she complained of epigastric pain that was alleviated with food ingestion. No nausea or vomiting was described.
The patient had a history of hypertension, dyslipidaemia, breast cancer in remission for 12 years and a hysterectomy 7 years previously. She was regularly medicated with captopril, simvastatin and aspirin.
At observation, the patient was afebrile and mildly hypertensive (BP 157/80 mmHg, HR 92 bpm) with no respiratory distress. She had bilateral basal crackles on pulmonary auscultation. There were no other relevant physical findings.
The diagnosis of heart failure in a patient with no known heart disease was assumed and chronic medication was optimized with the patient being started on a beta-blocker and loop diuretic. Additionally, she was started on omeprazole with resolution of the epigastric pain.
On follow-up, the patient remained asymptomatic for a year following the above medications. However, a year later the patient was admitted for decompensated heart failure and syncope. She had progressive worsening of dyspnoea, orthopnoea and bilateral pleural effusion.
The initial evaluation showed troponin I 1.39 µg/l, myoglobin 58 µg/l, N-terminal-pro BNP (NT-proBNP) 3,300 pg/ml, D-dimer 0.4 µg/ml, C-reactive protein 0.29 mg/dl and erythrocyte sedimentation rate 69 mm/1st h. Chest x-ray showed a normal cardio-thoracic index, and no effusions. The ECG showed sinus rhythm, HR 70 bpm, left axis deviation and low voltage in V2. The echocardiogram showed mild left ventricular concentric hypertrophy, preserved systolic function, with no motion changes, mitral moderate insufficiency III/VI and bi-atrial dilatation. An upper endoscopy revealed chronic gastritis (Helicobacter pylori negative).
During the following year, troponin and NT-proBNP values remained at a plateau (maximum troponin 1.69 µg/l and NT-proBNP 8620 pg/ml), while a repeat echocardiogram showed severe left ventricular concentric hypertrophy (ventricular septum thickness of 18 mm, posterior wall thickness of 16 mm). The coronary angiography did not show any lesions.
Holter monitoring performed after the syncope episode showed 294 sinus pauses (maximal R-R 5 sec).
Rapid progression of the left ventricular hypertrophy without ventricular dilatation, global diastolic dysfunction, with persistent elevation of troponin and NT-proBNP, low voltage on the ECG and the sinus dysfunction led to the diagnosis of restrictive cardiomyopathy, most likely cardiac amyloidosis.
A cardiac MRI showed a non-dilated left ventricle with marked concentric hypertrophy, especially in the septum (IVS 17 mm, LV mass 110 g/m2) and late enhancement in all portions of the left ventricle (yellow arrow in Fig. 1).
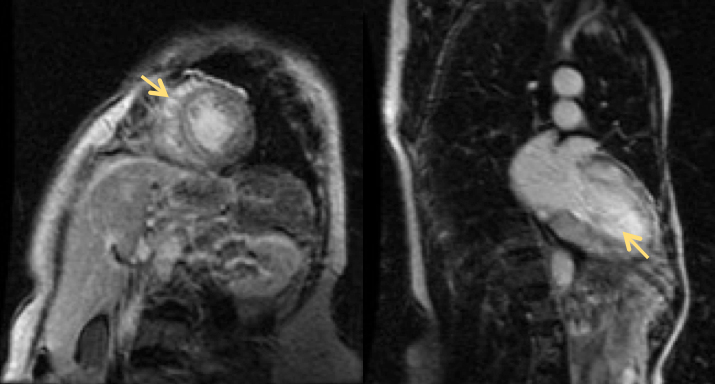
Figure 1 (click to enlarge)
Figure 1. Cardiac magnetic resonance imaging showing gadolinium late enhancement
The presence of amyloid substance was confirmed by an abdominal fat tissue biopsy. Bone marrow biopsy was normal and there was no increase in the free light chains in serum or urine, excluding primary amyloidosis. No other organ disease was identified, excluding systemic amyloidosis. The patient had no familiar history of amyloidosis. The diagnosis of SSA was therefore established.
The patient's medication was adjusted: heart failure was successfully managed with a loop diuretic (furosemide) and aldosterone antagonist (spironolactone), and after the ECG-Holter result, administration of beta-blocker was suspended. The patient had no other syncope episodes and a repeated ECG-Holter test showed a maximum R-R interval of 2 sec, so pacemaker implantation would not have been of benefit.
The patient returned asymptomatic to her daily routine activities and had regular Internal Medicine appointments.
Two years after her diagnosis the patient was admitted to the Internal Medicine ward with sudden clinical deterioration. She had congestive heart failure with asthenia, dyspnoea, severe anasarca, jugular venous distension and crackles on pulmonary auscultation. The ECG showed development of de novo atrial fibrillation. The patient was started on amiodarone for rhythm control and intravenous loop diuretics with no clinical improvement and steadily deteriorated until she died.
DISCUSSION
Amyloidosis is a rare disease characterized by the extracellular tissue deposition of protein fibrils which have an aberrantly folded beta-pleated sheet configuration. The type of protein precursor dictates which organ is affected.
The heart is the major organ involved in two amyloidosis subtypes: light chain amyloidosis and TTR amyloidosis (wild type TTR results in senile cardiac amyloidosis and mutated TTR in familial amyloidosis).
SSA is an acquired age-related amyloidosis. The protein precursor is wild-type TTR (non-mutant) and although the deposits can be found in numerous organs, the clinical manifestations are almost exclusively heart failure and carpal tunnel syndrome. The disease typically occurs in elderly men. Although wild-type TTR amyloid deposits can be found in a significant number of autopsies (in 50% of those over 90 years of age), only a minority will have clinically significant disease[1].
The predominant feature is infiltrative cardiomyopathy with associated progressive heart failure. The amyloid deposits cause thickened ventricular walls, impairing ventricular filling in the early phases and compromising systolic function in advanced stages. Diastolic dysfunction is the hallmark of the disease with dyspnoea, hepatomegaly and peripheral oedema being the main clinical features. Angina and syncope can also occur[2].
Syncope episodes have multiple causes and there are frequent conduction abnormalities that must be sought. Ventricular arrhythmias seem to be rare and cardioverter defibrillator implantation led to no improvement in preventing sudden death as most causes of sudden death are associated with non-shockable rhythm. In SSA, atrial fibrillation is seen in 10–15% of patients in advanced stages of the disease. The typical indolent progression of the disease can suddenly worsen due to the development of atrial fibrillation[2].
NT-proBNP and troponin are usually above the reference range in cardiac amyloidosis and are useful during the initial evaluation and follow-up.
ECG tracings show low voltage in the limb leads (≤0.5 mV) in 40% of cases and a pseudo-infarction pattern in 50% of them.
Echocardiography and cardiac magnetic resonance imaging (cMRI) are useful in the evaluation of these patients, but cannot differentiate the amyloid subtype and are not used as definite diagnostic tools. Nuclear studies, especially technetium 99mTC-based isotope imaging, are showing promising results in differentiating between light chain and TTR associated amyloidosis [3, 4].
The echocardiogram reveals symmetric concentric thickness of the left ventricular wall with associated diastolic dysfunction and restrictive physiology. A typical finding in the echocardiogram is increased echogenicity described as a sparkling appearance, which can be seen in 26–36% of patients[2]. The voltage-to-mass ratio allows the diagnosis of infiltrative cardiomyopathy; there is no real hypertrophy but there is increasing mass due to extracellular tissue deposits and therefore no high voltage on the ECG.
CMRI is particularly useful as a complementary diagnostic tool in identifying myocardial infiltration after the administration of contrast, and shows the distinctive pattern of left ventricle late gadolinium enhancement, a feature rarely seen in other cardiomyopathies.
Despite the previously described advances in imaging, a definite diagnosis is invasive and requires histological confirmation. Endomyocardial biopsy is an invasive procedure but is not mandatory; the diagnosis can be made in patients who have typical clinical findings and demonstrate amyloid deposits in samples from a non-cardiac site (e.g. rectal mucosa or abdominal fat tissue)[5].
SSA is a slowly progressive disease and no specific treatment is approved for this subtype. The aim is optimization of fluid status associated with heart failure symptoms. The mainstays of treatment are loop diuretics and aldosterone antagonists. Beta-blockers, angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists can aggravate the disease; negative inotropic calcium-channel antagonists are contraindicated[2]. In patients who develop atrial fibrillation, digoxin has increased toxicity potential; electric or pharmacological cardioversion must be considered[2, 3].
A new unexplained heart failure diagnosis, rapid disease progression with optimized therapy, increased ventricular thickness and particularly low voltage on the ECG must raise the suspicion of cardiac amyloidosis. The number of patients diagnosed with heart failure with preserved ejection fraction is increasing and some may have underdiagnosed infiltrative cardiomyopathy. SSA is a rare disease that presents as a common clinical syndrome but requires attention to its particular treatment and complications expected during the course of the disease.