ABSTRACT
Objectives: Infectious agents triggering haemophagocyticlymphohistiocytosis (HLH) primarily involve the herpes virus group. We report a case of HLH precipitated by Plasmodium falciparum.
Materials and methods: Clinical and laboratory findings in a patient presenting with fever were collected. After confirmation of acute malaria, anti-malarial treatment was administered.
Results: Despite initial favourable evolution, the patient developed fever again together with a worsening of the haematological parametersand increased ferritin levels. A bone marrow biopsy confirmed the diagnosis of HLH.
Conclusion: This case illustrates that HLH should be considered in the differential diagnosis of acute malaria in patients with persisting fever and pancytopenia.
LEARNING POINTS
- HLH may mimic infectious illnesses.
- HLH should be considered in the differential diagnosis ofacute malaria in patients with persisting fever and pancytopenia.
KEYWORDS
Plasmodium falciparum, hemophagocytic lymphohistiocytosis
INTRODUCTION
Haemophagocytic lymphohistiocytosis (HLH) is a clinical entitycharacterized by fever, pancytopenia and haemophagocytosis in bonemarrow and possibly other tissues[1]. It can occur as a primary syndrome, referredto as familial HLH, or as a syndrome secondary to an underlyingautoimmune, neoplastic or infectious disease, referred to as reactive HLH. Infectiousagents triggering reactive HLH primarily involve the herpes virus group,predominantly Epstein-Barr virus (EBV). Here we report a patient presentingwith reactive HLH precipitated by Plasmodium falciparum infection.
CASE REPORT
A 19-year-old Belgian male was admitted to hospital because of a fever(up to 40°C), which he had had for 2 days. He complained of headache,diffuse muscular pain and nausea. Four days before the onset of fever,he had returned from the Democratic Republic of Congo, where he hadstayed for 3 weeks. He had not taken malaria prophylaxis.
There was nothing of significance in his medical history.Physical examination showed a man who appeared ill, with a tympanictemperature of 39°C. His blood pressure was 135/74 mmHg and rate was 114 beats/min. Thecervical lymph nodes were slightly swollen. The rest of the physicalexam was normal.
Laboratory tests showed elevated levels of C-reactive protein and cytolytic liver enzymes as well as thrombocytopenia (Table 1). Chest X-ray was normal.
The clinical suspicion of acute malaria was confirmed by a thick blood smear, showing the presence of P. falciparum with only rare erythrocytes parasitized. A supportive treatment andoral quinine sulphate (500 mg tid), to be followed by doxycyclin fromday 3 onwards, was initiated. Because of gastrointestinal intolerance,this schedule was changed to atovaquone 250 mg/proguanil 100 mg, 4tablets daily for 3 consecutive days, starting on day 3. Abdominal ultrasound, performed on day 2of admission, showed slight splenomegaly (12.7 cm).
Although the clinical evolution was initially favourable, from day 3onwards, the patient started having a high fever again, up to 39°C. Hecomplained of headache and there was intermittent confusion with signsof meningismus. Gradual worsening of the haematological parameters wasobserved with deepening neutropenia and an increasing percentage ofatypical lymphocytes (Table 1).
| Day | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 8 | ||
| Maximum body temperature (°C) | 39.0 | 40.0 | 38.2 | 39.0 | 37.5 | 37.5 | 36.4 | 36.5 | |
| Normal Values | |||||||||
| Haemoglobin (g/dl) | 13.0-16.5 | 15.2 | 14.0 | 12.8 | 12.3 | 11.9 | 11.4 | 10.8 |
10.8 |
| Platelets (x103/mm3) | 158-450 | 79 | 52 | 30 | 27 | 43 | 50 | 84 | 194 |
| White blood cells (x103/mm3) |
3.6-9.6 | 3.5 | 3.7 | 1.5 | 1.8 | 3.8 | 3.9 | 4.5 | 6.7 |
| Neutrophils (x103/mm3) | 1.4-6.7 | 2.9 | 3.1 | 0.8 | 0.7 | 1.1 | 1.3 | 1.7 | 2.6 |
| Atypical lymphocytes (%) | 0.0 | 0.0 | 0.0 | 14.5 | 21.0 | 12.0 | 7.5 | 5.0 | 4.0 |
| Creatine kinase (U/l) | <145 | 288 | nd | 6604 | 5602 | 2250 | 813 | 466 | 205 |
| LDH (U/l) | 241-549 | 934 | nd | 3719 | 1934 | 1196 | 862 | 859 | 809 |
| ALT(U/l) | 21-72 | 111 | nd | 79 | 500 | 409 | 244 | 150 | 65 |
| AST (U/l) | 16-48 | 100 | nd | 592 | 430 | 366 | 288 | 235 | 146 |
| γ-GT (U/l) | 14-47 | nd | nd | 64 | 76 | 83 | 88 | 98 | nd |
| AF(U/l) | 65-220 | nd | nd | 110 | nd | 148 | 176 | 233 | 158 |
| Total bilirubin (mg/dl) | 0.2-1.0 | nd | nd | 4.94 | 4.83 | 3.93 | 2.67 | 2.10 | 1.59 |
| Direct bilirubin (mg/dl) | <0.5 | nd | nd | 3 | 3.2 | 2.3 | 1.6 | 1.1 | 0.6 |
| Fibrinogen (mg/dl) | 180-400 | nd | nd | 375 | 40 | 291 | |||
| CRP (mg/l) | <5 | 94.1 | 131.0 | 236.0 | 172.0 | 89.4 | 52.7 | 33.8 | 14.7 |
| PT (%) | >70 | 62 | nd | 68 | 76 | 87 | 94 | 91 | nd |
| Ferritin (μg/l) | 30-400 | nd | nd | nd | nd | 4102 | nd | nd | nd |
| Fasting triglycerides (mg/dl) | <150 | nd | nd | 467 | 727 | nd | nd | nd | nd |
LDH, lactate dehydrogenase; AST, aspartate aminotransferase; ALT, alanine aminotransferase; γ-GT, gamma-glutamyltransferase; AF, Alkaline posphatase or ALP; CRP, C-reactive protein; PT, prothrombin time; nd, not done
Table 1 - Evolution of tympanic temperature and laboratory parameters
New blood cultures remained negative. A thick blood smear showedpersistently low falciparum parasitaemia. Serologic studies werecompatible withpassed infection with EBV and cytomegalovirus. This clinical picture caused the suspicion of HLH. Triglyceridesand ferritin levels turned out to be strongly increased (Table 1).A bone marrow biopsy, performed on day 4, was normocellular withmegakaryocytic hyperplasia and a markedly increased number ofbenign-appearing histiocytes without nuclear atypia and exhibiting striking haemophagocytosis (Fig. 1).
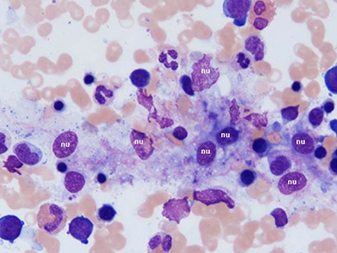
Figure 1a (click to enlarge)
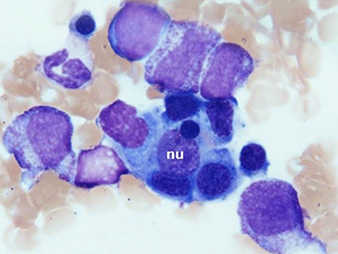
Figure 1b (click to enlarge)
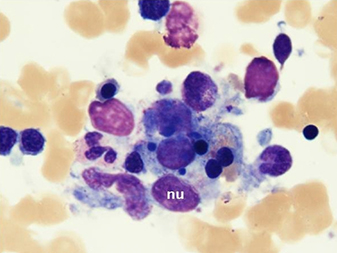
Figure 1c (click to enlarge)
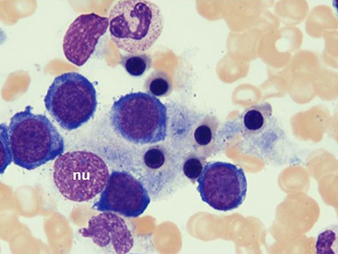
Figure 1d (click to enlarge)
Fig. 1 - Bone marrow biopsy
Bone marrow aspirate smears (May-Grünwald-Giemsa staining) demonstratemarked haemophagocytosis. Benign-appearing histiocytes contain small,round nuclei (nu) with condensed chromatin. The cytoplasm isabundant with ingestion of mature and immature haematopoietic elements,mainly erythroblasts.
According to the initial clinical evolution and the findingson blood smear, no other active treatment was considered.Broad-spectrum antibiotics (a combination of cefepime and amikacine intravenously) were givenas empirical coverage during febrile neutropenia.
From day 4 onwards, fever resolved and the haematological parameters began to improve (Table 1). Antibiotherapy was stopped on day 6 and on day 8 the patient left the hospital in a good, asymptomatic condition.
DISCUSSION
The term haemophagocytosis, first reported in 1939, describes thepathologic finding of activated monocytes, macrophages and histiocytesengulfing erythrocytes, leukocytes and platelets in bone marrow and othertissues[1,2].
Familial HLH is inherited in an autosomal recessive mannerwith symptoms usually evident within the first years of life[3]. Theassociation between HLH and infectious diseases can be challenging for the clinicianbecause both reactive and familial HLH are often precipitated byinfections. Moreover, HLH may mimic infectious illnesses, such as overwhelmingbacterial sepsis, or it may obscure the diagnosis of a precipitating,treatable infectious illness. Our patient developed signs of HLH within 2–3 daysafter the diagnosis of acute malaria. No other possible triggeringinfection could be demonstrated. HLH was suspected on clinical grounds, with adifferential diagnosis of acute malaria evolving into “severe malaria”or malaria complicated by bacterial septicaemia.
Although the underlying pathophysiologic mechanisms remainunclear, it is thought that all HLHs are due to some form of functionalimpairment of cytotoxic T lymphocytes and natural killer cells, resulting inthe inability to clear the antigenic stimulus and to turn off theinflammatory response, ultimately leading to hypercytokinaemia characteristic of HLH[4]. However, the exact mechanisms by which abnormal cytokineelaboration results in HLH are still elusive. Once the cytokine cascade has been triggered,HPS may continue to proceed independently of the presence of an initialtrigger.The diagnostic criteria for HLH include clinical, laboratoryand histopathologic features[5]. Fever and splenomegaly are the mostcommon clinical signs but hepatomegaly, lymphadenopathy, jaundice and rash havealso been described. Encephalopathy, meningismus and seizures are themost commonly reported central nervous manifestations. The most prominentlaboratory abnormality noted is cytopenia. Blood chemistry may suggesthaemolysis and most patients have hypertriglyceridaemia as well as marked elevationof serum ferritin. Serum fibrinogen is typically low.Histopathologically, haemophagocytosis can be demonstrated in bone marrow, spleen,liver and lymph nodes and occasionally in the central nervous system andthe skin. In our patient, HLH was suspected because of cytopenia affecting twolineages as well as prominently elevated serum ferritin and triglyceridelevels. A bone marrow smear allowed confirmation of the diagnosis.
For patients with reactive HLH associated with pathogens otherthan EBV, supportive care and treatment of the underlying infection isassociated with recovery in 60%–70%, as was the case in our patient[5].EBV-associated HLH is almost universally fatal if untreated. Therefore,patients should be treated with combination chemotherapy and immunotherapy (HLH-94treatment protocol), regardless of whether they are thought to havefamilial HLH. However, the distinction between reactive and familial HLHtriggered by a viral infection is important as allogeneic haematopoieticstem cell transplantation is the only curative therapy in patients withfamilial HLH who attain remission.
This case illustrates that HLH should be considered in thedifferential diagnosis of acute malaria patients with persisting feverand pancytopenia. Early recognition of this rare complication enables theclinician to consider timely immunosuppressive treatment in case thereis no spontaneous recovery and successful treatment of the underlying malariaattack alone.