ABSTRACT
Introduction: Isolated right pulmonary artery agenesis in an adult patient is an extremely rare condition that requires a high level of suspicion to make the diagnosis.
Case Description: A 32-year-old woman presented to the emergency room with a 4-month history of recurrent respiratory infections. Chest radiography and computerized tomography (CT) revealed alveolar opacities on the medium and inferior right lobes. Fibreoptic bronchoscopy with bronchial aspirate was negative on both cytological and microbiological analysis. Due to the persistent of the imaging findings after a full course of a wide-spectrum antibiotic, an angio-CT was performed, revealing a complete stop at the level of the right pulmonary artery. Angiography confirmed the diagnosis of right pulmonary artery agenesis.
Discussion: Currently, the patient has no exertional dyspnoea, screening for pulmonary hypertension has so far been negative and no further respiratory infections have occurred. It is important to call attention to a major congenital malformation that may remain asymptomatic until adulthood.
LEARNING POINTS
- Unilateral pulmonary agenesis is a rare entity that can present in multiple forms.
- A high level of suspicion and a thorough investigation is required for the diagnosis, with angiography remaining important even though chest angio-CT findings can suggest the diagnosis.
- IA major complication of this condition is pulmonary hypertension, which can appear early in infancy or with conditions that modify the pulmonary circulation such as pregnancy, although previous pregnancies did not trigger pulmonary hypertension in our patient.
KEYWORDS
Unilateral pulmonary artery agenesis, angiography
CASE DESCRIPTION
A 32-year-old woman presented to the emergency room complaining of persistent cough over the previous 4 months, relapsing fever (37.5ºC to 38ºC), right pleuritic chest pain and progressive dyspnoea. She had already received two courses of antibiotics. A chest radiograph and computerized tomography revealed alveolar opacities on the medium and inferior right lobes, with slight asymmetry in the dimensions of the lungs but no mediastinal abnormality (Fig. 1).
The patient was previously healthy with an uneventful medical history that included two pregnancies without any complications. She was a former smoker of 11 packs per year until the age of 29. She had an elevated C-reactive protein of 8.89 mg/dl (normal value <0.5 mg/dl) and mild hypoxemia (pO2 73 mmHg on room air), but all other blood tests were normal (including complete blood count and D-dimer). Blood cultures and serological tests (cytomegalovirus, Chlamydophila pneumoniae, Rickettsia, Mycoplasma, Coxsackie and Legionella pneumophila) were negative.
The patient was given a wide-spectrum antibiotic but nevertheless remained symptomatic for chest pain and exertional dyspnoea. We then extended the evaluation to include a serum angiotensin-converting enzyme, autoantibody screen which was also normal. Bronchoscopy showed inflammatory signs on superior and medium lobes on the right and mucous secretions. Both cytological and microbiological analyses of the bronchial aspirates were negative. Spirometry showed a mild restrictive syndrome, with a normal carbon monoxide transfer coefficient.
A new chest CT was performed which revealed persistence of the right lower lobe opacity and new surrounding ground glass opacities. This lesion was approached by surgical lung biopsy which revealed focal areas of lung infarction with no other significant morphological findings.
After this finding the patient underwent an angio-CT of the chest that revealed a complete stop at the level of the emergence of the right pulmonary artery with seemingly compensatory hypertrophied bronchial arteries to the right lung field (Figs. 2 and 3). The CT also showed that the right lung was slightly diminished in size and had reduced vascular markings, resulting in hyperlucency (Fig. 4). Angiography confirmed the diagnosis of right pulmonary artery agenesis (Fig. 5).
Currently the patient is completing 5 years of follow-up with mild exercise intolerance but with no other complaints. No further episodes of respiratory infection have occurred. Pulmonary hypertension has not developed so far. Pneumococcal vaccine was administered as well as annual influenza prophylaxis. The patient was advised not to get pregnant, and she agreed to tubal ligation. Clinical vigilance of the patient is maintained and a regular echocardiogram is performed in order to detect early signs of pulmonary hypertension.
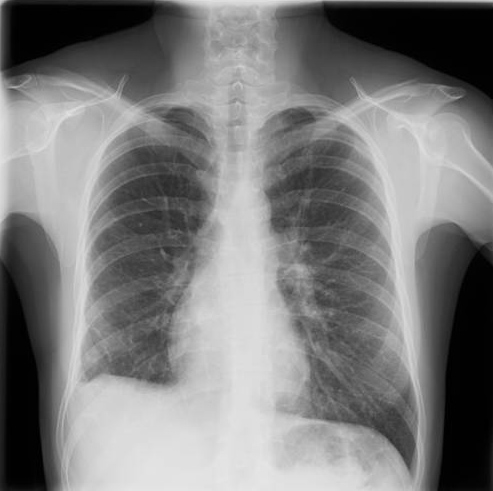
Figure 1 (click to enlarge)
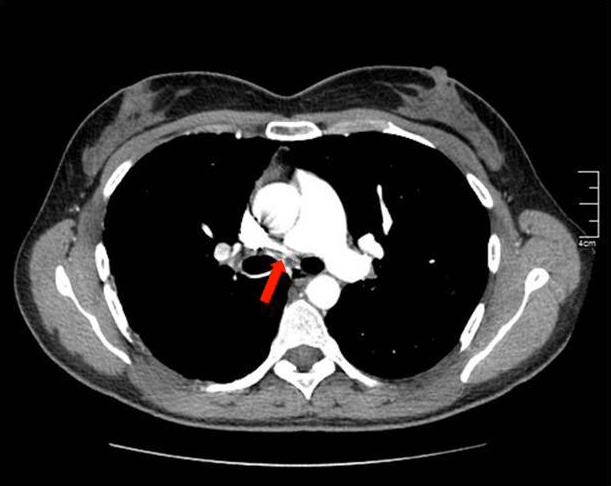
Figure 2 (click to enlarge)
Figure 1. Alveolar opacities on the medium and inferior right lobes, with slight asymmetry in the dimensions of both lungs
Figure 2. Pulmonary angio-CT revealing a complete stop at the level of the emergence of the right pulmonary artery (red arrow)
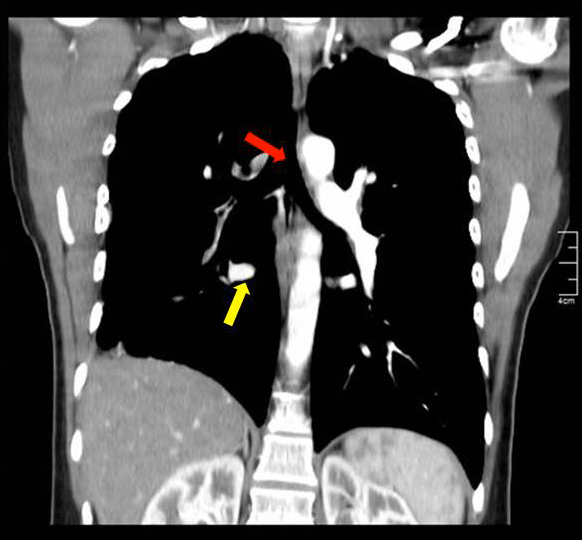
Figure 3 (click to enlarge)
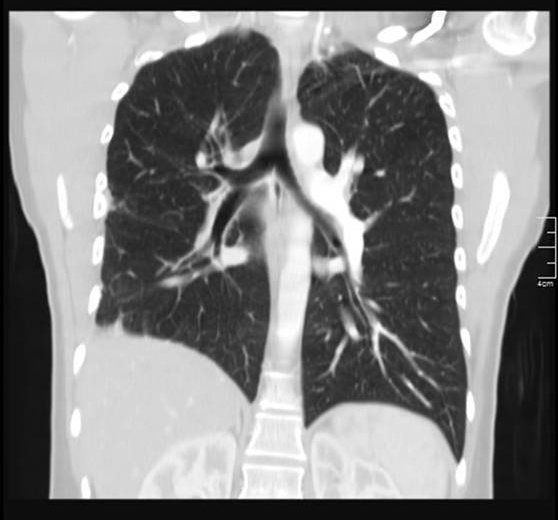
Figure 4 (click to enlarge)
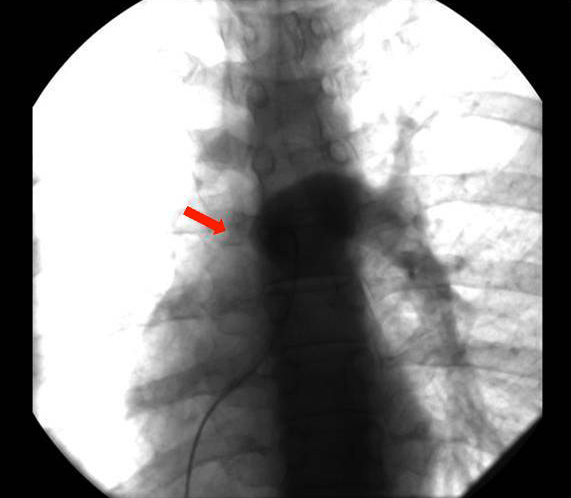
Figure 5 (click to enlarge)
Figure 3. Pulmonary angio-CT, coronal section, showing absence of the right pulmonary artery (red arrow), markedly enlarged bronchial arteries on the right hemithorax (yellow arrow) and reduced overall dimensions of the right lung
Figure 4 .Pulmonary angio-CT, coronal section, showing hyperlucency of the right hemithorax and reduced vascular markings of the right lung
Figure 5. Pulmonary angiography revealing total absence of the right pulmonary artery (red arrow) in agreement with the diagnosis of right pulmonary artery agenesis
DISCUSSION
Unilateral pulmonary artery agenesis (UPAA) is a rare entity with only a few hundred cases described in the literature[1]. The condition is usually associated with cardiac anomalies, but when isolated may be diagnosed in adulthood and the patient may remain asymptomatic[2]. The combination of clinical, laboratory and radiological findings, together with a high index of suspicion, are essential for diagnosis[2]. The incidence of UPAA is thought to be 1 in 200,000–300,000 adults, and is two thirds more frequent on the right[3].
There are two mechanisms for the agenesis: involution of the sixth aortic arch and persistence of the connection of the intrapulmonary arch to the distal sixth aortic arch, and closure of the ductus arteriosus after birth that causes loss of blood supply to the intrapulmonary pulmonary artery. In both cases systemic-to-pulmonary collateral arteries form[3]. These collaterals may arise from persistence of embryonic arteries, acquired collateralization from the bronchial, subclavian intercostal or diaphragmatic arteries or hyperplasia of the normal bronchial arteries[4].
In children, UPAA presents as pulmonary hypertension with right heart failure[3]. Most adults remain asymptomatic. The most common presentations in adulthood are haemoptysis, exercise intolerance and recurrent pulmonary infections[3]. Haemoptysis can present either in a mild but persistent form or as a massive haemorrhage that may require pneumonectomy or can lead to death[4]. Recurrent infection has a multifactorial aetiology. It may occur because of poor delivery of inflammatory cells to the affected lung, and ciliary malfunction. It may also occur due to alveolar hypocapnia as a result of poor blood supply, causing bronchoconstriction and trapping of mucous[4]. In adulthood it can also present as pulmonary hypertension, which occurs in 19–44% of patients as a result of the increased blood flow to the contralateral pulmonary artery, release of vasoconstrictive substances by the endothelium, and chronic vasoconstriction of the pulmonary arterioles. Half of patients have medial pulmonary artery hypertrophy in the normal lung. It is possible that patients who do not develop pulmonary hypertension early in life, are unlikely to develop it later[3].
The gold standard diagnostic method remains angiography[3], but with the advances in computed tomography and magnetic resonance imaging (MRI) it is now rarely performed. Usually the artery terminates 1 cm after the expected origin on the main pulmonary artery[3]. On CT or MRI, it is possible to find collateral circulation, an intact peripheral branch of the pulmonary artery and parenchymal changes due to recurrent infections[4]. Typically, on chest radiography there is a reduced lung field, a hyperlucent lung, an ipsilaterally shift of the mediastinum and a diminished hilar vasculature[4]. Transthoracic echocardiography is an important examination method as it can be used for evaluation of other congenital anomalies and pulmonary hypertension[4].
There is no consensus regarding treatment. In neonates and infants, surgical revascularization can be considered whenever feasible[5]. This is probably not possible in older patients as the intrapulmonary circulation of the affected lung is already fully established with a completely formed collateral circulation which is probably not suitable for surgical correction[3].
Remaining treatment for this condition is directed towards its possible complications. Pulmonary hypertension should be screened for and, if present, can be treated with vasodilators. Haemoptysis, if present, should be thoroughly evaluated and treatment can include direct embolization of the bleeding vessel, or in extreme circumstances, surgical approaches including pneumonectomy[3]. In some cases, recurrent pulmonary infections are treated with pneumonectomy[3].