ABSTRACT
Vogt–Koyanagi–Harada disease is rare, mediated by autoimmune melanocyte inflammation and facilitated by genetic predisposition[1-3]. The main clinical features include uveitis, meningitis, tinnitus and sensorineural deafness, and skin and hair depigmentation. It usually develops in four consecutive stages: prodromal, acute uveitic, convalescent, and chronic or recurrent[4]. In view of the first two stages, the differential diagnosis takes into account uveo-meningeal syndromes. Treatment is based on high dose corticosteroids. We present the case of a 14-year-old girl admitted to hospital with fever, progressive uveo-meningeal symptoms, and sensorineural hearing loss. After work-up, the final diagnosis of Vogt–Koyanagi–Harada disease was made.
LEARNING POINTS
- Vogt–Koyanagi–Harada disease should be considered when a patient presents with uveo-meningeal syndromes, particularly in pigmented ethnic groups.
- Acute bilateral uveitis should raise suspicion of Vogt–Koyanagi–Harada disease.
- Early stage diagnosis leads to prompt appropriate treatment and improves patient outcomes.
KEYWORDS
Vogt–Koyanagi–Harada disease, uveo-meningeal syndromes, aseptic meningitis
CASE DESCRIPTION
A 14-year-old girl was referred to the emergency department with a 72-hour history of fever and progressive weakness and malaise. Her past medical history did not reveal any remarkable symptoms, suggesting a viral infection. Born in Ecuador, South America, she was living in Spain, with no history of travelling outside the country for the previous 8 years. Any other epidemiologic circumstances, as well as family or personal past medical history, toxic habits, allergies or treatments were not relevant.
Physical examination showed a deteriorated general condition and weakness with normal osteo-tendon reflexes. Vital signs were the following: 32 breaths per minute, 101 heart beats per minute, BP 100/60 mmHg, temperature 39 ºC, and 96% base SpO2. A very slight unspecific rash was observed on both legs.
Emergency department laboratory work-up highlighted alanine transaminase 112 U/L (7-34); gamma-glutamyltransferase 141 U/L (4-24); C-reactive protein 150.3 mg/L (0.02-5); lactate dehydrogenase 415 U/L (0-247); haemoglobin 9.3 g/dL; haematocrit 27.4%; 80% neutrophils; 75,000 platelets; prothrombin time 17’’ (10.4-13), 59% (70-130); and activated partial thromboplastin time 50’’ (26-37). Chest X-ray showed a diffuse bilateral interstitial infiltrate. Urine sample and ECG were normal.
After admittance to our Internal Medicine Department, the course of the first week was characterized by evening mild fevers, with no determinant symptoms or signs. Complementary tests for systemic autoimmune, infectious and oncological diseases were negative or within normal values (Table 1).
On day 8 of admission, the patient developed acute blurred vision and meningeal symptoms. Examination of the pertinent fundus revealed an acute bilateral anterior uveitis (Fig. 1).
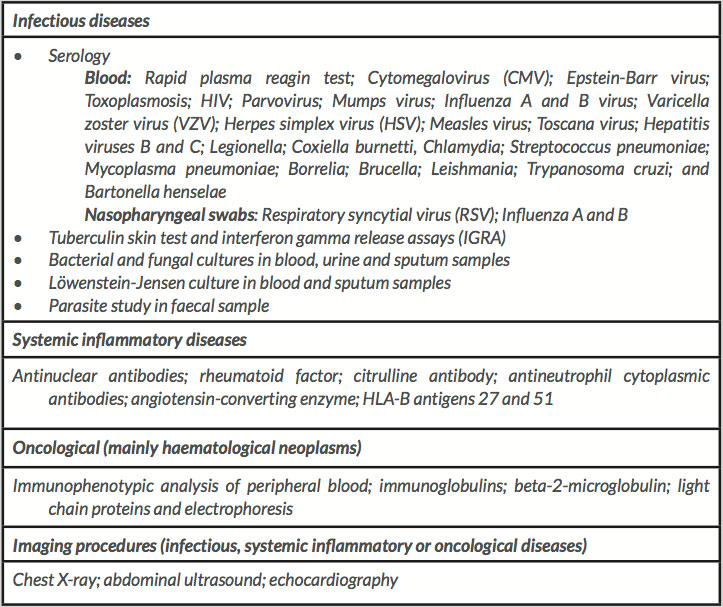
Table 1 (click to enlarge)
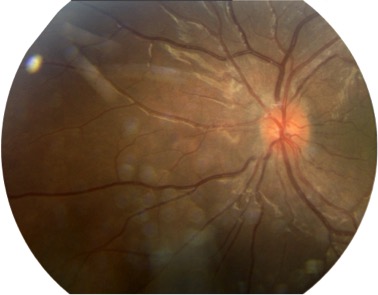
Figure 1 (click to enlarge)
Table 1. Complementary tests performed for differential diagnosis
Figure 1. Acute bilateral anterior uveitis
In addition, a lumbar puncture was performed showing normal pressure, normal glucose levels and lymphocytic pleocytosis in aseptic cerebrospinal fluid (CSF). Both findings were consistent with the diagnosis of uveo-meningeal syndrome, which led to further tests. Cranial and ocular nuclear magnetic resonance imaging, bronchoscopy and positron emission tomography showed no evidence of sarcoidosis, granulomatosis, infective or malignant lymphoproliferative disease, and the patient did not meet the diagnostic criteria of the International Workshop on Ocular Sarcoidosis.
On day 10 of admission, sudden hearing loss and tinnitus appeared. Audiometry showed a bilateral sensorineural deafness (Right: 45 dB, Left: 35 dB).
With these data – including uveo-meningeal syndrome and sensorineural hearing loss – and after ruling out any other infectious, systemic autoimmune, oncological diseases as well as any clinical evidence of Cogan syndrome, there was a high suspicion of Vogt–Koyanagi–Harada (VKH) disease. Based on the International Committee on Nomenclature for VKH and the positive test for HLA-DRB1 and HLA-DR4, the diagnosis of incomplete VKH disease was finally made.
In addition to the topical treatment for acute uveitis, oral prednisone (1 mg/kg/day: 45 mg/day) was initiated for 1 week, followed by a weekly tapering dose of 5 mg, until the dose of 35 mg (0.75 mg/kg) was reached and maintained for 6 months. Then, a new weekly tapering dose of 5 mg was initiated until its total discontinuation. The patient improved with complete resolution of symptoms, remaining asymptomatic after 1-year follow-up, without the appearance of skin or hair depigmentation.
DISCUSSION
VKH is an uncommon entity, affecting more women than men in the third to fourth decades of life, particularly in pigmented ethnic groups (Asian, Hispanic, Native American and Middle Eastern).
The accumulated evidence suggests that a viral environmental factor triggers in a genetically predisposed individual, as shown by the strong association with HLA antigens (DR4)[3], an autoimmune aggression against proteins of pigmented cells, due to molecular mimicry[1]. This leads to an aberrant inflammatory response affecting tissues and structures containing melanocytes[2] – eye, inner ear, central nervous system and skin – allowing for the clinical and pathogenic manifestations to develop into four phases:
I. Prodromal: This phase lasts 1 week and is clinically characterized by flu-like and meningeal symptoms (fever, headache, meningism). Its mayor sign is the pleocytosis in CSF.
II. Acute uveitic: This phase also lasts 1 week and is clinically characterized by acute blurred vision, hearing loss and tinnitus, along with acute bilateral uveitis (typically, posterior uveitis with exudative retinal detachments) and sensorineural deafness.
III. Convalescent: This phase can last for months or years and is characterized by asymptomatic skin and hair depigmentation, developing alopecia, poliosis, vitiligo and choroid depigmentation (sunset glow sign).
IV. Chronic or recurrent: This phase is characterized by complications of recurrent uveitis, atrophy and iris depigmentation[4]. The absence of this last stage establishes the denomination of incomplete VHK disease. Since this denomination generates doubts on the accuracy of the diagnosis and may delay the prompt instauration of treatment, thus compromising the outcomes, some authors have sought to change this denomination[5].
This case was challenging because of the large number of medical conditions associated with uveitis and uveo-meningeal syndromes as well as the rare entity that VKH is itself, but also because of the age of the patient and the unusual clinical presentation, especially concerning the atypical and late onset of uveitis.
Even though VKH disease is rarely observed in Europe, globalization and immigration of different ethnic groups convey uncommon diseases, of which physicians should be aware.