ABSTRACT
Objectives: To demonstrate difficulties in diagnosing and treating Addison’s disease caused by tuberculosis.
Materials and methods: We present a clinical case and review of the literature.
Results: A 62-year-old man presented with gastrointestinal symptoms, weight loss and enlarged adrenal glands. After 2 months of diagnostic tests, a working diagnosis of Addison’s disease due to extrapulmonary tuberculosis was made. Treatment was challenging due to interaction between rifampicin and steroids.
Conclusion: Our case illustrates that in non-endemic countries, extrapulmonary tuberculosis still needs to be considered as a possible cause of Addison’s disease.
LEARNING POINTS
- In non-endemic countries, extrapulmonary tuberculosis still needs to be considered as a possible cause of Addison’s disease.
- Treating tuberculosis and adrenal cortex insufficiency can be challenging because of the interaction between rifampicin and adrenocorticoid drugs.
- Adrenal function does not recover in most cases of Addison’s disease caused by tuberculosis.
KEYWORDS
Tuberculosis, Addison’s disease, adrenalitis
INTRODUCTION
When Thomas Addison described his patients with adrenal cortex insufficiency in 1855, all cases were due to destruction of the adrenal cortex by Mycobacterium tuberculosis[1]. The incidence of tuberculosis (TB) has since declined in the Western world and TB is no longer the most common cause of Addison’s disease (AD). Given the often non-specific symptoms of AD and the low incidence of TB in non-endemic countries, the diagnosis is easily overlooked and/or delayed. Experience in diagnosing TB is becoming rare, resulting in a higher frequency of misdiagnosis in recent years[2]. We present a case of AD caused by tuberculous adrenalitis discovered in the diagnostic work-up of gastrointestinal symptoms. The case demonstrates the difficulties in identifying the diagnosis and establishing a definite TB diagnosis. In addition, it illustrates treatment difficulties due to drug interactions.
CASE REPORT
A 62-year-old man was admitted to hospital for analysis of weight loss, nausea and loss of appetite without fever. His medical history included sleep apnoea, anaemia due to blood donations and a previous suspicion of ocular sarcoidosis. On examination, hyperpigmentation of the skin and a blood pressure of 87/62 mmHg were found. Blood tests revealed hyponatremia (131 mmol/l), normal potassium (4.4 mmol/l), a decrease in kidney function (eGFR MDRD 47 ml/min) and increased inflammation parameters (CRP 16 mg/l, ESR 52 mm/h) with normal leukocyte count (5×109/l) and differential. A CT scan showed bilateral enlargement of the adrenal glands (Fig. 1) and cervical, thoracic and abdominal lymphadenopathy. Cortisol levels indicated primary adrenocortical failure (basal cortisol 62 nmol/l, ACTH 72 nmol/l) and hydrocortisone and fludrocortisone were started. TB was considered the most likely cause, other considerations being lymphoma or metastases of an unknown primary tumour. It subsequently became clear that the patient had been born in Indonesia and had stayed in a TB clinic in the Netherlands at the age of eight. An attempt to obtain lymph node material by surgical exploration of the cervical region and axillary ultrasound-guided biopsy was unsuccessful. Subsequent adrenal biopsy specimens showed necrosis, infiltration of histiocytes and granulomas. Auramine staining of the first specimen revealed acid-fast bacteria, the microscopic features consistent with M. tuberculosis bacilli (Fig. 2).
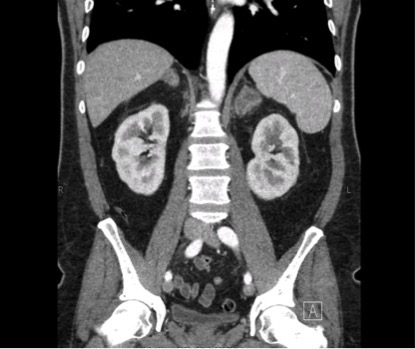
Figure 1 (click to enlarge)
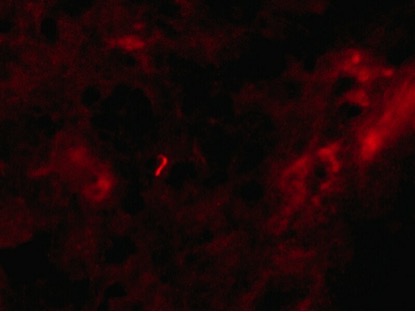
Figure 2 (click to enlarge)
Figure 1. CT scan of the abdomen with enlarged adrenal glands (frontal view).
Figure 2. Acid-fast bacillus in the first adrenal biopsy specimen after auramine staining
PCR was inhibited, culture was negative. In the second specimen, PCR and culture were both negative and no bacilli were found. Enzyme-Linked ImmunoSpot (ELISPOT) on blood and brochoalveolar lavage fluid was positive for M. tuberculosis, while culture and PCR were negative. A working diagnosis of AD caused by adrenal TB was made and the patient was put on a course of tuberculostatic medication consisting of isoniazid, rifampicin, pyrazinamide and ethambutol. Doses of hydrocortisone and fludrocortisone were increased two- to threefold because of interaction between rifampicin and steroids. After 2 months of treatment, a CT scan showed decreased lymphadenopathy and a reduction in the size of both adrenal glands. Symptoms resolved and 1 year after presentation, the patient is still on hydrocortisone, fludrocortisone and tuberculostatics.
DISCUSSION
This case illustrates the difficulties in recognizing and treating a severe disease that presents with common symptoms. The patient, presenting with gastrointestinal symptoms and weight loss, had AD due to destruction of the adrenal glands by TB infection. Although cervical, thoracic and abdominal lymph nodes were enlarged, TB infection of other organs could not be established. Hormone substitution dosage had to be adjusted because of interaction with tuberculostatic medication. Adrenal function did not recover after 1 year of TB treatment.
The adrenal glands are a common site of extrapulmonary TB. Tuberculous AD is caused by destruction of the adrenal glands by caseous necrosis. At least 90% of the adrenal gland has to be destroyed before adrenal insufficiency occurs. Therefore, adrenal function is unlikely to recover after treatment. The adrenal glands are often (70%) bilaterally involved, which is the result of haematogenous or lymphogenous spread from the primary site of infection[3]. In a Japanese study, 93% of patients with tuberculous AD had experienced non-adrenal TB in the past[4]. Nevertheless, it is not uncommon for the adrenal glands to be the only infected organ: in a large autopsy study, the adrenal glands were the only site of infection in 25% of patients with adrenal TB[3].
Concurrent treatment of AD and TB can be challenging as rifampicin is a strong inducer of the cytochrome P450 (CYP) system, involved in the metabolism of adrenocorticoids. This interaction results in lower plasma levels of steroid drugs, which can be potentially dangerous. No exact guidelines for dose adjustment are available, but it is advised that the dose should be adjusted when the enzyme inducer is started and stopped[5]. We increased the dose of steroid drugs three- (hydrocortisone) and twofold (fludrocortisone) on initiating treatment with tuberculostatics.
In retrospect, presentation with a history of previous TB in combination with clinical signs and symptoms of AD and enlarged adrenal glands raised a high suspicion for tuberculous adrenalitis in our case. Still it took almost 2 months and several invasive procedures before a working diagnosis was made and treatment started.
Diagnosing AD can be challenging due to the non-specificity of presenting symptoms. When only weight loss and gastrointestinal symptoms are the main complaints, AD will not feature highly in the differential diagnosis. Timely recognition of the disorder is important because of the risk of an adrenal crisis. In our case, recognizing AD as the most likely cause of symptoms only occurred after the bilateral adrenal enlargement was observed on a CT scan. Given the signs and symptoms of this patient, AD could have been considered earlier and cortisol levels could have been measured before radiological imaging was ordered.
In addition, several invasive procedures were performed in an attempt to establish a definite TB diagnosis by culture of M. tuberculosis and/or PCR. In the end, the combination of pathological findings and the positive auramine staining on the adrenal biopsy in conjunction with the overall presentation was deemed sufficient to clinically establish the diagnosis and start therapy. One might argue that the clinical signs in combination with enlarged adrenals and a positive ELISPOT on blood might have been enough, precluding further invasive procedures.
In conclusion, AD caused by tuberculous adrenalitis presents with non-specific symptoms and onset is often insidious. Diagnosis is therefore frequently overlooked, enhancing the risk of an adrenal crisis. Treating the infection and adrenal cortex insufficiency can be challenging because of interaction between rifampicin and adrenocorticoid drugs. Adrenal function does not recover in most cases of AD caused by tuberculosis.