ABSTRACT
Acute anaemia is characterized by a reduction in the number of red blood cells, haemoglobin levels or haematocrit. By far the most common aetiology is haemorrhage, but in its absence other less frequent causes should be considered.
The authors present the case of a 42-year-old man with a diagnosis of glucose-6-phosphate deficiency and progressing gastric signet ring cell carcinoma, who was admitted to the internal medicine department for symptomatic back pain control. During his hospitalization, the patient developed acute anaemia with schistocytes on peripheral blood smear with no concurrent hyperbilirubinaemia or decreased haptoglobin. Bone metastatic disease was documented. The case was revised with the transfusion medicine department and malignancy-associated microangiopathic haemolytic anaemia (Ma-MAHA) was suggested. The patient was transferred to the oncology department and later discharged, dying at home shortly afterwards.
LEARNING POINTS
- The possibility of multiple aetiologies for anaemia in the same patient should be considered.
- Pathophysiological mechanisms are important in the differential diagnosis of anaemia.
- Malignancy-associated microangiopathic haemolytic anaemia can present as a paraneoplastic syndrome.
KEYWORDS
Acute anaemia, paraneoplastic syndrome, schistocytes, thrombotic microangiopathies
CASE REPORT
A 42-year-old man with a past medical history of glucose-6-phosphate dehydrogenase deficiency was diagnosed with gastric signet ring cell carcinoma (cT3N1M0). The patient underwent perioperative chemotherapy with the MAGIC protocol (epirubicin, cisplatin, fluorouracil) and total gastrectomy but disease progression was documented soon afterwards (ypT3N3bM0). One year after the diagnosis, the patient presented to the emergency department with non-mechanical dorso-lumbar pain, radiating to the right lower limb, with radicular paraesthesias. Blood tests revealed macrocytic normochromic anaemia with Hb 10.1 g/dl, VGM 100 fl, leucocytes 9,200×109/l, thrombocytopenia of 59,000×109/l and LDH 303 U/l. A plain dorso-lumbar vertebral radiograph and posterior CT scan showed no signs of metastasis or of radicular compromise.
Physical examination on admission showed a slim patient, with normal vital signs and cardiopulmonary auscultation. There was a positive Lasègue sign on the right lower limb at 20º elevation, but no other relevant findings. On the 7th day of hospitalization, the patient developed a fever (38.6ºC), maintaining haemodynamic stability with no signs or symptoms suggestive of active infection. There was no jaundice, choluria, neurological symptoms or skin changes apart from paleness of mucous membranes. Routine blood tests revealed an acute drop in haemoglobin (Hb) level to 4.7 g/dl, with no visible haemorrhage or signs of internal bleeding, and a reticulocyte index of 1.7. A blood smear requested by the transfusion medicine department revealed schistocytes, thrombocytopenia of 43,000×109/l, LDH 724 U/l, a normal white blood count, INR 1.2, hypofibrinogenaemia (3.51 mg/dl) and increased haptoglobin (314 mg/dl). Bilirubin, urea and creatinine levels were normal. Due to suspicion of a nosocomial infection, blood and urine cultures were taken, serology tests for HIV, HBV, HCV, CMV and EBV were carried out, and broad-spectrum antibiotics were started (Table 1).
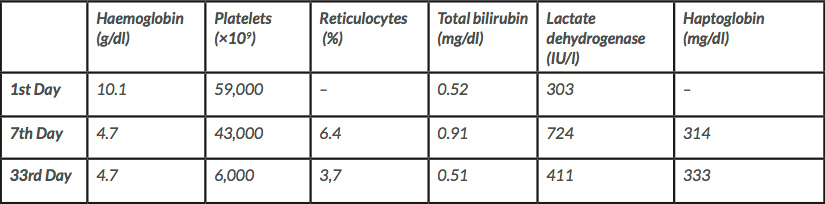
Figure 1 (click to enlarge)
Table 1. Evolution of biochemical markers of haemolysis during the patient’s hospital stay
Medication was revised on the hypothesis that it could have triggered favism exacerbation and bone marrow aplasia, but no suspicious drugs were found. Bone marrow biopsy was postponed after discussion with the haematology department as it would not change the therapeutic approach due to worsening of the patient’s performance status. During the remaining course, the patient was given RBC transfusions with no significant increase in Hb values. Antibiotics were suspended after negative blood and urine cultures, and negative serology for the above mentioned viruses. The patient was started on 1 mg/kg corticosteroids, with no laboratory response, prompting suspension. Bone scintigraphy later showed diffuse bone metastases (Fig. 1). The case was discussed with the oncology department to decide whether to start chemotherapy, but after evaluation the patient was considered ineligible. He was later transferred to the oncology department and was discharged after 50 days of hospitalization and Hb values persistently below 8 g/dl despite RBC transfusions when symptomatic. He died at home 3 days later with the continuous support of a palliative care team.
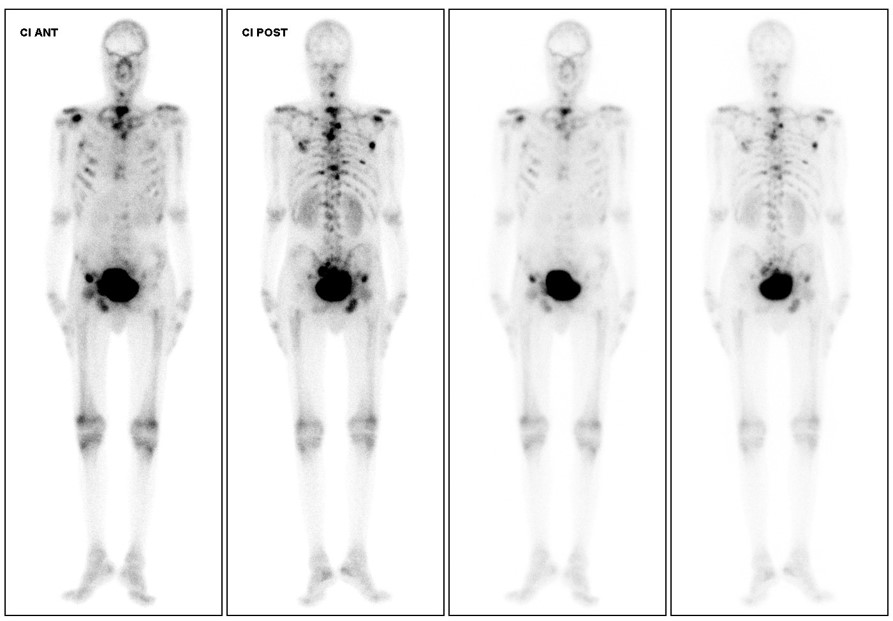
Figure 1 (click to enlarge)
Figure 1. Bone scintigraphy showing axial and peripheral bone metastasis
DISCUSSION
After acute haemorrhage was excluded due to the lack of a relevant history and absence of physical findings, the hypothesis was a hypoproliferative anaemia or an acute haemolytic anaemia. The presence of schistocytes suggested intravascular haemolysis, but our case had some important differences compared with the literature.
Acute exacerbation of favism was considered but excluded because it typically shows the classic laboratory signs of haemolysis, absence of Heinz bodies and bite cells on peripheral smear, which were absent in this case.
A thrombotic microangiopathy was considered in the differential diagnosis. The diagnosis of thrombotic thrombocytopenic purpura (TTP) requires the presence of microangiopathic haemolytic anaemia (MAHA) and thrombocytopenia, with or without neurological or renal abnormalities, and the absence of an alternative aetiology[1]. Even though the clinical presentation of TTP may vary, our patient never had neurological, renal or dermatological changes that would support such a diagnosis. Neither did he have the changes in the coagulation profile that are common in this disease, and so disseminated intravascular coagulation was ruled out. TTP is usually associated with a poor short-term prognosis (patients typically die within 1–2 weeks), but our patient survived for more than 30 days after the acute event, which does not fit the disease profile. Systemic malignancies can mimic the diagnostic features of TTP, but there is often a history of malignant disorder, or there are atypical features that make the diagnosis of TTP less certain[1].
Malignancy-associated MAHA (Ma-MAHA) is a rare paraneoplastic syndrome which is more frequent in patients with gastric tumours, followed by breast, prostate and lung cancers[2]. It is usually associated with metastatic disease and with poorer outcomes[2,3]. Although there was no biochemical evidence of haemolysis, the presence of schistocytes suggested intravascular destruction of red blood cells, and so the authors consider haemolysis may have contributed to the aetiology of this patient’s anaemia. We hypothesize there was low grade haemolysis, which is supported by the presence of schistocytes, which would explain the lack of hyperbilirubinaemia as the liver was functioning normally. With progressive bone marrow infiltration, a hypoproliferative medulla could not compensate for the small daily changes in haemoglobin level, culminating in the low level found. The presence of slightly elevated levels of haptoglobin could be explained by it being an acute phase protein, elevated either by an infection or by the pro-inflammatory state that the cancer itself confers. The Ma-MAHA reports in the literature describe a disease course more like that of our case than that of patients with TTP.
Regarding possible therapeutic approaches in this particular case, the removal of immune complexes by plasmapheresis was discussed with the transfusion medicine department but discarded due to insufficient evidence of benefit in Ma-MAHA[1,2]. Chemotherapy, which is the only therapy with proven benefit in Ma-MAHA[1], was also considered, but the patient’s performance status was too low.
We consider it important to share this case because despite the work-up results, it is difficult to determine a specific aetiology. Our case of acute anaemia was probably multifactorial in origin, with contributions from hypoproliferation due to bone marrow infiltration by cancer cells documented by bone scintigraphy, Ma-MAHA, and a precipitating event such as an acute nosocomial infection or drug interaction, which could not be confirmed. It was a diagnostic challenge as there was a wide range of possible causes, some of which were uncommon, and superimposed characteristics; we felt that reviewing the pathophysiological phenomenon of these diseases helped to guide our diagnostic work-up. We would also like to point out that early recognition of MAHA, as a paraneoplastic syndrome, can improve prognosis, with chemotherapy being the only appropriate treatment[1].