ABSTRACT
Pheochromocytoma, a rare neuroendocrine tumour, is often encountered in the general internal medicine clinic as an adrenal incidentaloma. Even rarer is its presence in adult cyanotic heart disease, although there are a few documented reports of this association in various paediatric populations, with chronic hypoxia being the likely driving force. Here we present the case of a 38-year-old adult with unrepaired complex cyanotic congenital heart disease with biochemically proven pheochromocytoma presenting as an adrenal incidentaloma to a general internal medicine clinic.
LEARNING POINTS
- There is an association between pheochromocytoma and congenital cyanotic heart disease.
- It is important to maintain clinical vigilance for laboratory testing for pheochromocytoma in patients with cyanotic heart disease who are seen at general internal medicine clinics.
KEYWORDS
Pheochromocytoma, cyanotic heart disease
INTRODUCTION
Adrenal incidentalomas and congenital heart disease (CHD are commonly encountered in general internal medicine (GIM) clinics, but their co-existence is uncommon.
Pheochromocytoma is a rare neuroendocrine tumour which accounts for about 5% of the adrenal incidentalomas presenting with or without sympathetic symptoms such as palpitations, uncontrolled hypertension and anxiety[1,2]. There are some reports in the literature describing an association between pheochromocytoma and cyanotic CHD, which is thought to be secondary to chronic hypoxia[3]. There are also a few case reports describing this association in the literature but they mostly refer to the paediatric population[1–5]. Here we present the case of an adult with unrepaired complex cyanotic CHD with biochemically proven pheochromocytoma presenting as an adrenal incidentaloma to a GIM clinic.
CASE DESCRIPTION
A 38-year-old man was seen at the GIM clinic with non-specific abdominal symptoms such as pain, bloating and constipation. His comorbid conditions included unrepaired cyanotic CHD, secondary polycythaemia due to resting hypoxia, hyperuricemia and latent TB. His medications included Lasix, allopurinol, ranitidine and aspirin. He was a non-smoker, non-drinker, and had no family history of cardiac disease or adrenal tumours or paragangliomas.
His underlying unrepaired cardiac anatomy showed congenital transposition of the normal position of the great arteries of the heart, severe pulmonary stenosis, a double inlet and double outlet right ventricle (RV) with mild RV dysfunction, a hypoplastic left ventricle (LV) connected to the RV by a large ventricular septal defect (VSD), and a transposed aorta anterior to the right of the pulmonary artery with moderate aortic valve regurgitation. His CHD was diagnosed at birth in Iraq and was managed conservatively with medication. He had baseline symptoms of NYHA class 4 dyspnoea and intermittent chest pain with severe functional limitation throughout his adult life. He was awaiting cardiac surgery to improve his overall status.
He presented to the GIM clinic with acute abdominal symptoms such as bloating, constipation and nausea of unclear aetiology. In the work-up of his abdominal symptoms, a CT of the abdomen was ordered which revealed an incidental right adrenal mass measuring 2.5×2.1 cm at 41 Hounsfield units. The rest of his work-up was normal. Further exploration revealed a long-standing history of palpitations, anxiety and tinnitus on a background of severe PTSD from war experiences. He has also needed phlebotomies for secondary polycythaemia due to ongoing hypoxia as he requires 5 l/min of oxygen at rest. His physical examination was remarkable for hypoxia on room air, blood pressure of 110/60 mmHg, heart rate of 70, finger clubbing and a single S1/S2 with a grade 3/6 systolic ejection murmur at the 3rd–4th left lower sternal border.
An initial laboratory investigation included a 24-hour urine cortisol, metanephrines and catecholamines, which showed significantly elevated norepinephrine and normetanephrine (Table 1). His other investigations were all normal and included CT of the chest, ANA, AMA, hepatitis serology, IgG, IgA, IgM, SPEP, LFTs, ferritin, TSH and extended electrolytes.
Subsequently, an MIBG scan with planar and SPECT imaging was performed with 370 mBq I-123. The MIBG scan showed prominent uptake in the adrenal glands with the right less than the left without any metastasis (Fig. 1). Given the degree of uptake in the left and right adrenals along with the positive biochemical results, the diagnosis of pheochromocytoma was made. The patient was subsequently started on an alpha blocker and referred to a specialized cardiac anaesthesia team for possible resection of the tumour.

Table 1 (click to enlarge)
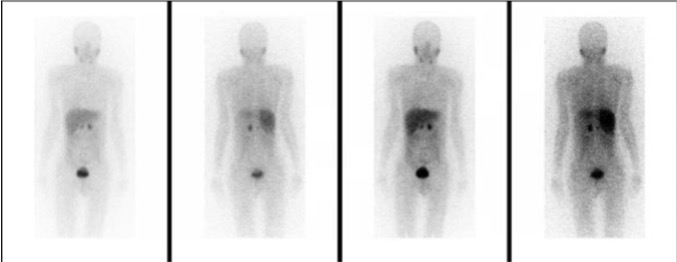
Figure 1(click to enlarge)
Table 1. Laboratory investigations
Figure 1. MIBG scan showing increased uptake in the adrenal glands
DISCUSSION
Pheochromocytoma and paragangliomas are rare neuroendocrine tumours arising from neural crest-derived cells such as the adrenal gland, and often presenting to the general medicine clinic as incidentalomas[1,2] Even more uncommon are these tumours in patients with cyanotic CHD, as in our patient. However, there are a few reports of this association, mostly in the paediatric population[3–5].
Regarding the association between these entities, some evidence points to a link between the chronic hypoxia pathway and genetic neuroendocrine tumour syndromes. Pheochromocytoma is associated with germline mutations in the RET proto-oncogene, and the von Hippel-Lindau (VHL), neurofibromatosis-1 (NF1) and succinate dehydrogenase complex (SDH) genes[1–3]. As some of the protein products of these genes, namely VHL, mediate the cellular response to hypoxia by activating the hypoxia-inducible factor (HIF) signalling pathway, it is plausible that hypoxia is the main driver of tumour growth[1–3]. The fact that our patient had been hypoxic since birth supports the above theory. A recent observational study showed an association between these entities, but the time course of hypoxaemia was not well established[3]. Polycythaemia can be a secondary manifestation of cyanotic heart disease but can also be seen in pheochromocytoma alone.
Given the increased survival of patients with CHD into adulthood, patients presenting for non-cardiac surgery are posing unique medical and surgical challenges. Initial treatment is with an alpha-blocker together with management by a cardiologist specializing in CHD. There are also specific anaesthetic management techniques for resection of pheochromocytomas in adulthood, which is usually carried out at a specialized centre[6,7].
In summary, this case highlights the association between these two rare entities, and the importance of clinical vigilance for laboratory testing for pheochromocytoma in patients with CHD who are seen at GIM clinics. Although hypoxia is the link between the two entities, this factor has not yet been extrapolated to other chronic cyanotic diseases such as end-stage chronic obstructive pulmonary disease or interstitial lung disease, which could be an area of future research.