EJCRIM 2023 CiteScore
| 2.1 = | 1.730 Cit. to date |
| 842 Docs. to date |
Last updated on 05 April, 2024
Updated monthly
Updated monthly
Powered by



|
Views: 1584
HTML: 300
PDF: 518
Supplementary file: 0
Untitled: 0
Untitled: 0
|
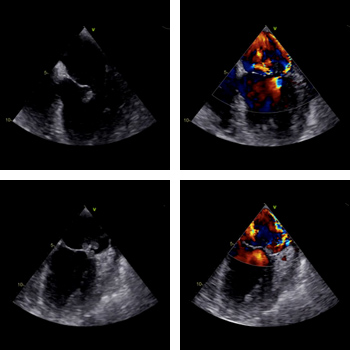
Infective endocarditis (IE) is associated with high morbidity and mortality despite advances in antibiotic and surgical treatment. Systemic embolism occurs in up to 49% of IE patients and may involve the major arteries, limb arteries, viscera and the central nervous system. In this report we describe a 60-year-old female patient with a history of acute lymphoblastic leukaemia who presented with endocarditis manifesting as stroke, acute limb ischaemia and meningitis. Early diagnosis is essential since treatment lowers the risk of embolism, with most events occurring within 2 weeks of treatment initiation.
|
Views: 1143
HTML: 701
PDF: 489
Cover letter: 0
Figures, tables, consent: 0
|
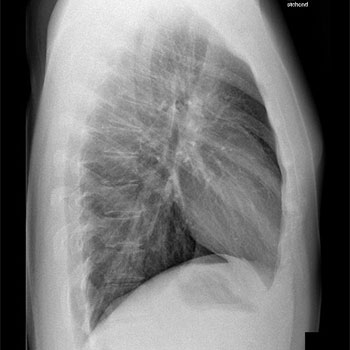
The case of a 25-year-old expedition doctor who developed high altitude pulmonary oedema (HAPE) while climbing in the Swiss Alps is presented, with reference to the literature. The patient’s symptoms of HAPE were typical. Less typical was the fact that the doctor had previously been to similar altitudes uneventfully. The only differentiator is that on this expedition he developed a mild lower respiratory tract infection (LRTI) 2 days prior to travel. There has been limited, conflicting evidence regarding LRTI as a risk factor for HAPE and high quality research has not focused on this area. LRTI is not commonly recognised as being a risk in high altitude environments, which may be resulting in lethal consequences. This report aims to inform, provide a clinical question for future high altitude research expeditions, and encourage consideration by expedition and high altitude doctors.
|
Views: 1287
HTML: 498
PDF: 474
Figure 1: 0
Figure 2: 0
Figure 3: 0
|
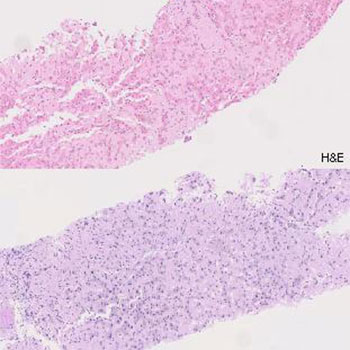
Light-chain deposition disease (LCDD) is a rare monoclonal gammopathy that involves the deposition of light chains (LC) in multiple organs, leading to progressive dysfunction. The kidney is usually the most affected organ and responsible for the initial clinical manifestations. We present the case of a patient with LCDD with prominent liver involvement (marked cholestasis, hepatomegaly and portal hypertension) but with no evidence of coexisting lymphoproliferative disorder.
|
Views: 1201
HTML: 1482
PDF: 531
Figure 3: 0
Figure 1: 0
Figure 2b: 0
Figure 2: 0
|
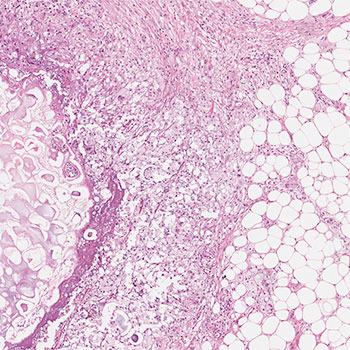
Pancreatic panniculitis is a rare disorder affecting 2–3% of patients with pancreatic disease. The findings are characterized by tender, erythematous, subcutaneous nodules which may undergo spontaneous ulceration with discharge of brownish and viscous material derived from colliquative necrosis of adipocytes. The lesions are usually localized in the lower limbs, although they may also extend to the buttocks and also involve the trunk, upper limbs and scalp. They can precede overt pancreatic disease in 40% of cases. The typical histological features observed in these lesions are characterized by necrotic adipocytes with absent nuclei (better known as ‘ghost cells’) in the context of a predominantly lobular panniculitis.
We describe the case of a 78-year-old cirrhotic woman admitted to our department with abdominal pain affecting the upper abdomen and a 3-day fever. On physical examination, multiple tender erythematous nodules, with irregular margins, were present on the pretibial regions of both lower legs, ranging in size from 0.8 to 1.5 cm. Pancreatic amylase and lipase were elevated and abdominal computed tomography revealed acute pancreatitis with oedema, focal gland enlargement of the pancreatic tail and perivisceral inflammation. Histological examination of the lesions was consistent with a diagnosis of necrotizing granulomatous panniculitis.
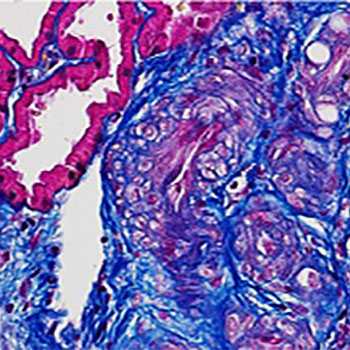
|
Views: 3747
HTML: 278
PDF: 456
Figure 2: 0
Letter of submission: 0
Copyright: 0
Figure 1: 0
|
Antiphospholipid syndrome (APS) is an autoimmune disease which can be primary or secondary to other autoimmune conditions and is defined by the occurrence of arterial or venous thrombosis, or pregnancy morbidity associated with persistently positive antiphospholipid antibodies (aPLA). The kidney may be affected by thrombosis at any level of its vasculature. When small vessels are involved, this results in thrombotic microangiopathy (TMA), which can manifest as either acute vaso-occlusive or chronic vascular lesions in glomeruli, arterioles and interlobular arteries. We report the case of 26-year-old man, with a previous medical history suggestive of APS, who was found to have a small elevation in serum creatinine. A kidney biopsy was performed and revealed features of chronic TMA. Anticoagulation was begun and kidney function remained stable. However, one year later, upon suspension of anticoagulation, the patient developed acute kidney injury and a second kidney biopsy showed acute TMA. This case describes different manifestations of antiphospholipid syndrome nephropathy (APSN) and highlights the importance of anticoagulation for thrombosis prevention.
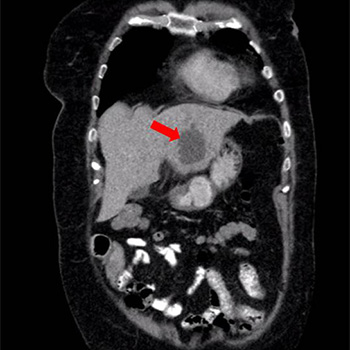
|
Views: 1085
HTML: 445
PDF: 391
Untitled: 0
|
Pyogenic liver abscess is a potentially devastating clinical entity associated with significant morbidity and mortality[1]. A myriad of causes for liver abscess have been described including intra-abdominal infections such as diverticulitis[2]. Due to a non-specific presentation, clinicians often require a high level of suspicion in their diagnosis of this condition. A handful of cases of liver abscess have been described following colonoscopy which was usually a complicated procedure or one where multiple biopsies had been taken[3,4]. The case of a patient presenting pyrexia of unknown origin one week after undergoing an uncomplicated colonoscopy in which no biopsies were taken is reported. She was ultimately diagnosed with a pyogenic liver abscess.
|
Views: 1321
HTML: 650
PDF: 487
PSH Assessment Measure: 0
|
Patients who survive a traumatic brain injury (TBI) can sometimes experience symptoms of excessive sympathetic discharge. Despite being known about for more than 60 years, the diagnostic criteria for this condition have only recently been defined under the name "paroxysmal sympathetic hyperactivity". Failure to recognize this syndrome leads to excessive costs, prolonged hospital stays and delayed recovery for TBI patients. This case report describes a patient whose specific rehabilitation program was affected by a failure to identify this entity, even though he presented with many of the characteristics of this condition.
|
Views: 2319
HTML: 3173
PDF: 456
Skin lesions compatible with Schambers´s disease: 0
|
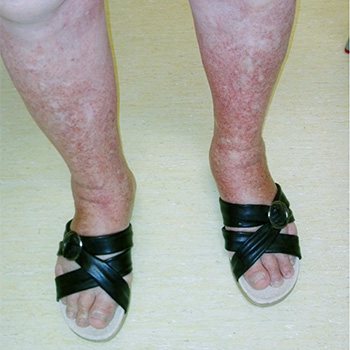
Pigmented purpuric dermatosis is a chronic benign skin disorder of unknown aetiology. Although there are several other potential cofactors, drugs are the most frequent cause. This paper describes the case of a 71-year-old woman who was admitted in the emergency department with skin lesions on the lower extremities, characteristics of Schamberg's disease. After a medical study and treatment, it was concluded that the lesions were caused by amlodipine administration. To the authors’ knowledge, only one previous case describing an association between this disease and amlodipine administration has been reported in the medical literature.
| 2.1 = | 1.730 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2023, Published by SMC Media srl, Italy - Privacy policy

