EJCRIM 2023 CiteScore
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |
Last updated on 05 May, 2024
Updated monthly
Updated monthly
Powered by



|
Views: 256
HTML: 13
PDF: 152
|
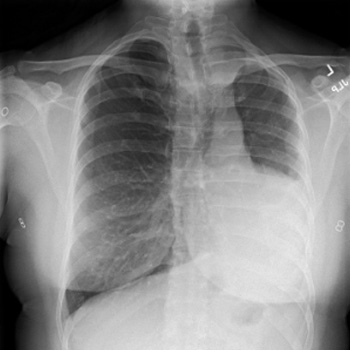
Background: Alagille syndrome (ALGS) is a multisystem disorder involving at least three systems among the liver, heart, skeleton, face, and eyes. Common cardiac associations include pulmonary artery stenosis/atresia, atrial septal defect (ASD), ventricular septal defect (VSD) and tetralogy of fallot (ToF). Coarctation of aorta (CoA), renal and intracranial arteries are commonly involved vessels in Alagille syndrome. We present two cases with rare cardiovascular manifestations of Alagille syndrome.
Case description: Case 1: A 25-year-old female with a history of Alagille syndrome presented to the cardiologist office for progressive exertional dyspnoea, orthopnoea, and palpitations. She was tachycardiac on examination and had an apical diastolic rumble. A transthoracic echocardiogram (TTE) showed a left ventricular ejection fraction (LVEF) of 60% and parachute mitral valve (PMV) with severe mitral stenosis. A transoesophageal echocardiogram (TOE) showed insertion of chordae into the anterolateral papillary muscle, severe mitral stenosis with a valve area of 0.7 cm. She was referred to a congenital heart disease specialist and underwent robotic mitral valve replacement with improvement in her symptoms.
Case 2: A 27-year-old female with known Alagille syndrome and resistant hypertension presented to the cardiologist office due to progressive exertional dyspnoea for a year. She was hypertensive and had a new 2/6 systolic ejection murmur along the left upper sternal border. TTE revealed an LVEF of 60% and pulmonary artery pressure of 19 mmHg. A CoA was suspected distal to the left subclavian artery due to a peak gradient of 38 mmHg. Cardiac magnetic resonance (CMR) imaging ruled out CoA, and diffuse narrowing of the descending thoracic aorta measuring 13–14 mm in diameter was noted. The patient was referred to a congenital heart disease specialist for further management.
Conclusion: PMV presenting as mitral stenosis and mid-aortic syndrome are not commonly described anomalies in association with Alagille syndrome. TTE, TOE and CMR played a key role in diagnosis and management of these patients.
|
Views: 138
HTML: 16
PDF: 162
|
Lung underdevelopment is a rare congenital anomaly with variable clinical significance and presenting symptoms. It usually manifests during childhood. We present two cases of developmental lung anomaly subtypes and discuss clinical presentation and outcomes in such patient populations.
|
Views: 270
HTML: 124
PDF: 249
|
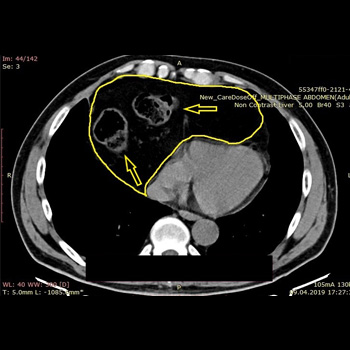
Background: Congenital diaphragmatic hernias are rare congenital defects resulting in abdominal organ protrusion into the thoracic cavity; they mainly present with pulmonary or gastrointestinal symptoms. Although congenital and discovered in utero or in early childhood, they can be asymptomatic for a long time and even remain asymptomatic despite the growing hernia sac dimensions and the hernia sac contents.
Case description: We present a case of a 58-year-old patient with incidentally diagnosed Morgagni hernia during the COVID-19 pandemic following a computerised tomography (CT) scan of the chest. He presented without any symptoms related to the existence of the hernia. Another CT scan was performed 20 months after the initial diagnosis to evaluate the progression of the hernia. The patient refused the offered surgery due to the absence of symptoms.
Discussion: A Morgagni hernia is usually discovered during pregnancy or in early childhood, but sometimes can be asymptomatic for years. Main symptoms originate from the respiratory and gastrointestinal system.
Conclusion: Due to the refusal of surgery, we were able to follow the CT scan enlargement progression of patients’ hernia over a 20-month period.
|
Views: 496
HTML: 62
PDF: 383
|
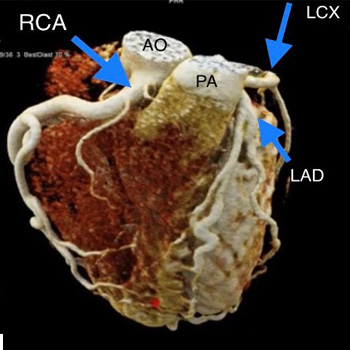
Anomalous left coronary artery from the pulmonary artery (ALCAPA) is considered a rare congenital heart disease where the take-off of the left coronary artery abnormally originates from the pulmonary artery instead of left aortic sinus. It is associated with a high mortality rate in the first year of life and sudden death in adults if left untreated. We report an adult form of ALCAPA syndrome in a 20-year-old female who presented with anginal pain for the previous few months. Unfortunately, the patient was hesitant to have surgery at the time.

|
Views: 432
HTML: 78
PDF: 512
|
Background: Hereditary angioedema is a rare hereditary and potentially life-threatening disorder characterized by recurrent attacks of cutaneous and submucosal swelling. In spite of the advances made in terms of pathophysiology, underlying mechanisms are not fully clear and this, in turn, hinders the development of effective therapies. Currently, on demand treatment is considered first-class, with few cost-effective, long-term prophylactic options.
Case presentation: Here we describe the case of a 34-year-old man diagnosed with hereditary angioedema at the age of 10, who used to suffer several angioedema attacks per month. He was given prophylactic treatment with antifibrinolytic agents and androgens without improvement. Moreover, he was treated with plasma-derived C1-INH concentrate or icatibant for on-demand treatment of moderate and severe angioedema attacks. At the age of 33, after suffering sudden vision loss and lower limb paresthesia, he was studied and diagnosed with multiple sclerosis. Teriflunomide was administered at a dosage of 14 mg/day. Angioedema attacks disappeared 40 days after starting treatment.
Conclusion: Thus, we suggest considering the pathophysiologic mechanisms on which teriflunomide could be active and consider this drug carefully as an option for prophylaxis purposes. Yet, its effectiveness on this condition should be further studied.
|
Views: 220
HTML: 83
PDF: 264
|
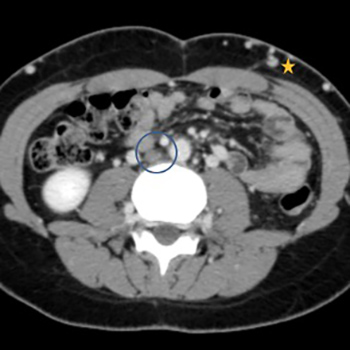
Biliary hamartomas or von Meyenburg complexes (VMCs) are hepatic tumour-like lesions related to congenital malformation of the ductal plate, and are part of the ciliopathy spectrum of disorders. The exact pathogenesis of VMCs is unclear and it remains controversial whether they have the potential for malignant transformation. Patients are often asymptomatic and VMCs are usually encountered as an incidental finding on imaging.
We report a case of recurrent sepsis with an unidentified focus. It was later confirmed that biliary hamartomas were acting as a sanctuary for the persistent pathogenic agent. The authors hope to draw attention to the existence of this unusual focus of recurrent sepsis.
|
Views: 430
HTML: 118
PDF: 350
|
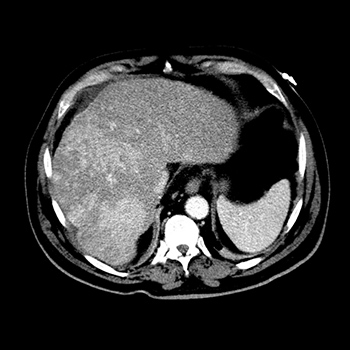
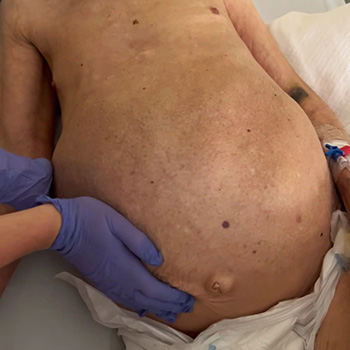
Caroli disease is a rare congenital pathology caused by mutation of the PKHD1 gene (polycystic kidney and hepatic disease 1), also responsible for autosomal recessive polycystic kidney disease. Characterized by segmental and multifocal dilatation of the large intrahepatic bile ducts, classic disease involves only malformation of the biliary tract. The association with congenital hepatic fibrosis is called Caroli syndrome. We describe the case of an 84-year-old man with Caroli syndrome diagnosed in 1997 by liver biopsy. The CT scan revealed massive hepatomegaly, extending to the pelvic region, and almost total replacement of the parenchyma by numerous cystic formations, no evidence of bile duct dilatation, and no ascites or splenomegaly suggestive of portal hypertension. The atypical clinical presentation, with no reported complications, resembles that of a space-occupying lesion with an indolent course, previously misdiagnosed as metastatic neoplasm.
|
Views: 362
HTML: 107
PDF: 308
|
Classical Ehlers-Danlos syndrome (cEDS) is one of the 13 subtypes of Ehlers-Danlos syndrome, which has the major clinical criteria of hyperextensibility skin, atrophic scars, and generalised joint hypermobility. The occurrence of aortic dissection has been described in some subtypes of Ehlers-Danlos, but it has a rare association with the cEDS subtype. This case report discusses a 39-year-old female with a past medical history of transposition of great arteries with a Senning repair at the age of 18 months and controlled hypertension with medication, who presents a spontaneous distal aortic dissection. The diagnosis of cEDS was made using the major criteria, and a novel frameshift mutation in COL5A1 was discovered. The reported case emphasises that in patients with cEDS, vascular fragility may be a complication.
|
Views: 324
HTML: 57
PDF: 331
|
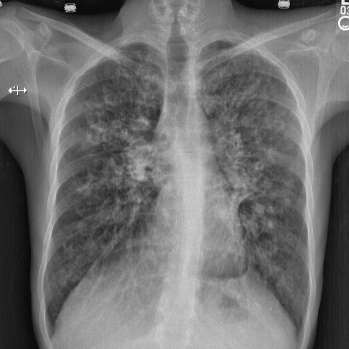
Cystic fibrosis (CF) is a common autosomal recessive disorder which is mainly found in Caucasians but has also been reported in Asian populations. CF is primarily caused by mutations in the CFTR gene which regulates the transport of chloride ions across the cell membrane. We describe the cases of two siblings with CF diagnosed with the rare missense mutation c.80G>T, which has only been referenced once in the literature and shows a possible association with classical form of CF.
|
Views: 580
HTML: 64
PDF: 328
|
Inferior vena cava (IVC) atresia is a rare congenital vascular malformation. We describe the case of a 20-year-old woman with IVC atresia who presented with a 3-month history of fatigue, oedema of the lower limbs and episodes of lipothymia. Transthoracic echocardiography and cardiac catheterization were performed, revealing interruption of the IVC with circulation through the azygos and hemiazygos system. An abdominal and pelvic computerized tomography (CT) scan confirmed the findings, demonstrating the absence of the IVC below the renal veins. Blood tests did not reveal any relevant results. These findings are consistent with the diagnosis of IVC atresia, a rare condition with no standard treatment. As a surgical approach was not possible, pharmacological measures were implemented for primary prevention of possible thrombotic events.
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2024, Published by SMC Media srl, Italy - Privacy policy

