EJCRIM 2023 CiteScore
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |
Last updated on 05 May, 2024
Updated monthly
Updated monthly
Powered by


|
Views: 337
HTML: 55
PDF: 252
|
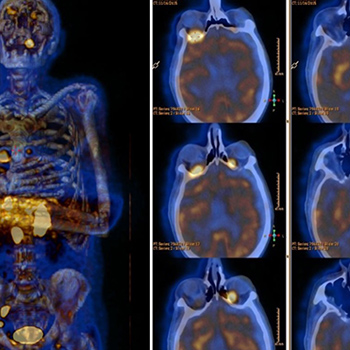
Intraocular lymphoma (IOL) is a rare and life-threatening condition whose aetiology is unclear. Blurred vision, reduced vision, and floaters are common initial symptoms. Posterior vitreous detachment and haemorrhage rarely occur.
The authors present the case of a 79-year-old man who initially presented with a 3-month history of fever, night sweats, significant weight loss, bilateral peri-orbital haematoma, red eyes and retro-orbital headache. Physical examination revealed fever, bilateral peri-orbital haematoma, subconjunctival haemorrhage and palpable cervical lymphadenopathy. CT scans detected conical intra-orbital lesions, cervical adenomegalies, expansive lesions in the adrenal glands, and thrombosis of the splenomesenteric confluent and posterior segment of the right branch of the portal vein. These findings were suggestive of a lymphoproliferative disorder. Aspiration cytology of the adrenal mass and inguinal adenopathies was compatible with diffuse large B-cell lymphoma with areas of transformation to Burkitt’s lymphoma.
We describe a rare form of lymphoma, and a very unusual presentation of primary intraocular lymphoma with atypical symptoms.
|
Views: 351
HTML: 55
PDF: 316
|
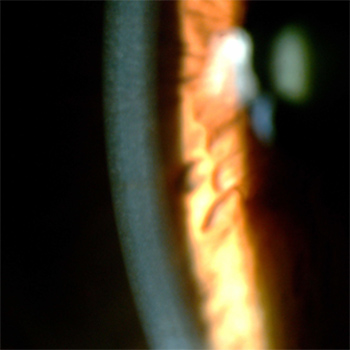
We report the case of a 70-year-old man diagnosed with late-onset Wilson disease (WD) with mild neurological symptoms only and a new mutation in the ATP7B gene. A compound mutation of the ATP7B gene was found with the variant c.98T>C p(Met33Thr) in exon 2, in heterozygosis, and variant c.2224G>A (Val742Ile) in exon 8, in heterozygosis. Patient age should not be a determinant for excluding WD. Genetic sequencing is an important tool for the discovery of new genetic mutations.
|
Views: 446
HTML: 55
PDF: 294
|
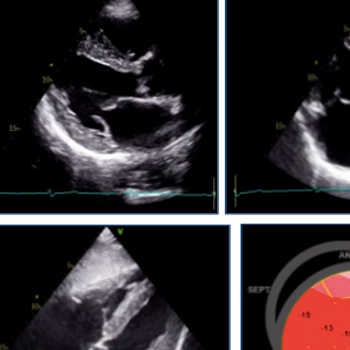
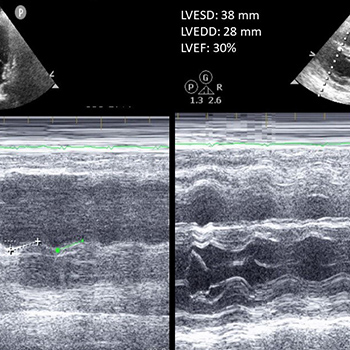
A 60-year-old man, with a history of familial lipodystrophy, hypertriglyceridaemia, hepatic steatosis and bone cysts, was admitted due an acute coronary event. Coronary angiography showed significant stenosis in the left anterior descending artery, which was treated. Transthoracic echocardiography showed a slightly dilated left ventricle with diffuse and heterogeneous thickening of its walls, slightly decreased left ventricular function and reduced global longitudinal strain. Due to these echocardiographic findings, cardiac magnetic resonance imaging was requested, which identified intramyocardial diffuse fibrosis of the basal septum and points of insertion of the left and right ventricles, without oedema, microvascular obstruction or myocardial infarction. Owing to the constellation of symptoms and distinctive features on cardiac imaging, a diagnosis of Berardinelli-Seip congenital lipodystrophy (BSCL) was suspected, which was confirmed through genetic testing of the pathogenic variants in BSCL2 and AGPAT2. BSCL is a rare autosomal recessive syndrome characterized by the congenital absence of adipose tissue and triglyceride deposition in other tissues, such as muscle, liver and heart.

|
Views: 476
HTML: 58
PDF: 328
|
Takayasu arteritis is a systemic vasculitis of the large vessels and mainly affects Japanese and Southeast Asian women in the second and third decades of life. Inflammatory infiltrate affects the full thickness of the vessel wall, inducing progressive lumen stenosis and occlusion. The main biomarkers of disease activity are the ESR, CRP and serum levels of circulating cytokines.
This case report describes the clinical history of a young woman with Takayasu disease with high serum levels of IgA at onset. IgA remained elevated with persistence of disease activity, and normalized only when the patient was treated with an anti-TNF agent (infliximab), which also induced a clinical response in the vasculitis.
IgA levels, together with other inflammatory parameters, may be considered a biomarker of disease activity.
|
Views: 373
HTML: 59
PDF: 252
|
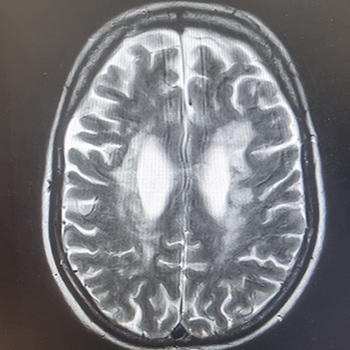
A woman with recurrent thromboembolic stroke was found to also have cor triatriatum. When the patient first presented with weakness, she was thought to have ischaemic stroke because she had conventional risk factors, but she was later confirmed to have cor triatriatum. The main method of treatment is surgery. However, if surgery is contraindicated, anticoagulation can be used as second-line treatment, but this can be difficult. This report describes the follow-up of a middle-aged female patient with cor triatriatum over 6 years during which she experienced multiple strokes despite different methods of anticoagulation.

|
Views: 534
HTML: 67
PDF: 333
|
ntroduction: Non-episodic angioedema associated with eosinophilia (NEAE) has been reported primarily in young East Asian women and is characterized by a single episode of persistent limb oedema, peripheral eosinophilia, and transient joint pain. Although there are reports of eosinophilia disease after coronavirus disease 2019 (COVID-19), the occurrence of NEAE has not been previously reported.
Case description: A 29-year-old Japanese woman, with a history of allergic rhinitis and atopic dermatitis, sought a medical consultation for persisting oedema of the extremities, which developed about 2 weeks after she contracted COVID-19. Physical examination revealed symmetrical non-pitting oedema with peripheral predominance. Laboratory examination revealed a blood eosinophil count of 7536/µl. The patient was diagnosed with NEAE and a 7-day course of prednisolone (15 mg/day) was initiated, with rapid improvement in the oedema and no recurrence on follow-up.
Discussion: The exact aetiology of NEAE is unknown, but it may develop after infection or drug exposure. Eosinophilic disease after COVID-19 infection has been reported and, therefore, eosinophilic angioedema should be considered in the differential diagnosis of non-pitting oedema of the extremities after a COVID-19 infection. Early diagnosis of NEAE is important as rapid improvement can be achieved with low-dose steroid treatment.
Conclusion: NEAE can develop after COVID-19 and should be considered in the differential diagnosis of non-pitting oedema of the extremities.

|
Views: 575
HTML: 97
PDF: 467
|
We report the case of a 31-year-old woman with a history suggestive of obstetric antiphospholipid syndrome (APS) with recurrent miscarriages, preterm labour and intrauterine fetal death. During her last pregnancy, she was referred to the Rheumatology Clinic at King Fahad Military Medical Complex, Dhahran, Saudi Arabia. Serology for connective tissue diseases and APS was negative on multiple occasions. During previous pregnancies, her obstetrician had initiated several trials of baby aspirin with and without prophylactic heparin, without success. We diagnosed her with seronegative obstetric APS (SN-APS). A specific regimen, consisting of combination therapy with baby aspirin, low-molecular-weight heparin, hydroxychloroquine (<5 mg/kg/day) and low-dose prednisolone, was attempted. She delivered a healthy baby even though it was born preterm at 30 weeks of gestation because of abruptio placentae. Obstetric SN-APS is rare and should be considered and, if the history is highly suggestive, treated similarly to seropositive obstetric APS to reduce mortality.
|
Views: 403
HTML: 112
PDF: 254
|
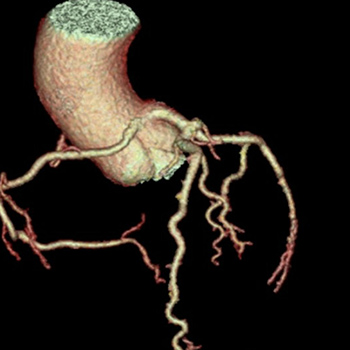
Patients with symptomatic or malignant anomalous aortic origin of the right coronary artery (AAORCA) warrant surgical treatment to decrease morbidity and mortality. Various surgical techniques have been implemented including unroofing, reimplantation and bypass grafting. A 43-year-old woman presented with intermittent chest pain due to malignant AAORCA and received saphenous bypass grafting, instead of reimplantation, due to intraoperative spasm.
|
Views: 442
HTML: 66
PDF: 303
|
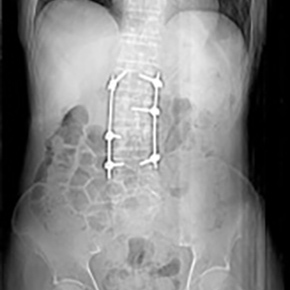
Coxiella burnetii causes Q fever, which is found worldwide and can be acute or chronic. This case report describes a 72-year-old man whose bilateral lower limb pain revealed a paravertebral abscess at L2–L3 due to Q fever spondylodiscitis. Surgical drainage of the abscess was performed and medical treatment is ongoing.
Q fever is endemic in Portugal and transmitted by inhalation of aerosols containing spores from infected animals (cattle, goats and sheep) or by ingesting cottage cheese or unpasteurized milk. It has an incubation period of 2–3 weeks and 60% of patients are asymptomatic with only 2% needing hospitalization. Primary infection can manifest in any organ and most cases are self-limiting (self-limited febrile illness, atypical pneumonia or acute hepatitis). Less than 1% of cases evolve to chronic disease, presenting as osteomyelitis or endocarditis.
Chronic disease poses a diagnostic challenge and spondylodiscitis has an insidious evolution. Diagnosis requires microbiological and clinical confirmation. Serological and polymerase chain reaction tests are used for diagnosis. <br/>
Acute disease is usually treated with doxycycline for 3 weeks to avoid evolution to chronic disease. Chronic disease requires 18–24 months of doxycycline with hydroxychloroquine. Acute disease can recur so follow-up is essential as chronic Q fever can result in morbidity and mortality. In Portugal Q fever is a notifiable disease due to the epidemiological risk.

|
Views: 303
HTML: 176
PDF: 281
|
Gaucher disease (GD) is a rare, autosomal recessive genetic disease caused by deficiency of a lysosomal enzyme (glucocerebrosidase and B-glucosidase) that leads to the accumulation of its substrate in lysosomal macrophages. GD remains rare and delayed diagnosis is common due its gradual onset. It is important to include this differential diagnosis in cases of massive splenomegaly and/or thrombocytopenia, in order to avoid potentially harmful splenectomy.
This case report describes a 25 year old female patient with a 10 year medical history of anaemia and thrombocytopenia, who presented with symptoms of haemorrhagic dyscrasia, pancytopenia and massive splenomegaly. The differential diagnosis of massive splenomegaly included several conditions which were considered but ruled out. Because of a lack of resources, the patient was forwarded to a reference centre where the diagnosis of GD was made
|
Views: 544
HTML: 58
PDF: 382
|
Denosumab is one of the most commonly used antiresorptive drugs for osteoporosis treatment and the prevention of skeletal-related events in cancer patients. The purpose of this case report is to highlight potentially life-threatening severe hypocalcaemia as a side effect of denosumab complicated by refractory shock that failed to respond to medical management including intravenous calcium, vasopressors and inotropes in an elderly man with a history of prostatic cancer.
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2024, Published by SMC Media srl, Italy - Privacy policy

