EJCRIM 2023 CiteScore
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |
Last updated on 05 May, 2024
Updated monthly
Updated monthly
Powered by


|
Views: 963
HTML: 266
PDF: 597
|
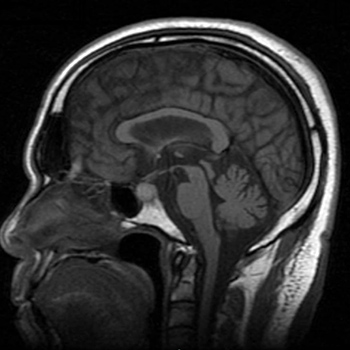
Pituitary apoplexy is a rare endocrine emergency, characterized by a sudden increase in pituitary gland volume secondary to acute ischaemic infarction or haemorrhage of the pituitary gland, usually in the presence of a pituitary adenoma. We present the case of a 79-year-old man admitted for new-onset, bi-temporal and severe headache, associated with photophobia and vomiting, whose additional study revealed pituitary apoplexy. This case highlights the need for high clinical suspicion of this rare entity in order to reduce the associated mortality.
|
Views: 1115
HTML: 85
PDF: 492
|
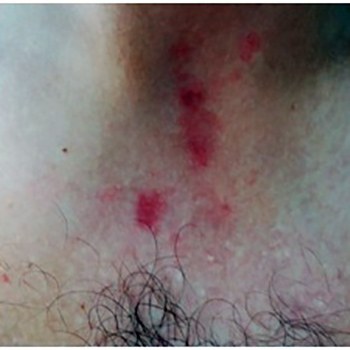
Terlipressin is used for the treatment of bleeding oesophageal varices and hepatorenal syndrome in patients with cirrhosis. Adverse effects are usually minimal. However, potentially serious side effects such as skin necrosis involving the extremities, scrotum, trunk and abdominal skin can rarely occur. Our patient had greater skin involvement than other described cases. We present the case of a patient with extensive skin necrosis, unrelated to the infusion site, in the lower and upper limbs, scrotum and abdomen after the use of terlipressin. Skin necrosis secondary to terlipressin is a rare complication and early identification is essential so the drug can be immediately suspended.
|
Views: 1145
HTML: 92
PDF: 438
|
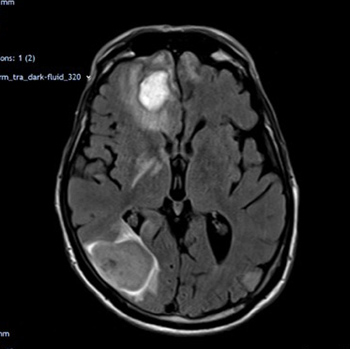
The aim of this case report and literature review is to explore the rarely highlighted co-occurrence of malignant melanoma and Parkinson’s disease (PD) and to describe the devastating effects on those affected. We present a case of acute confusion and worsening shuffling gait in a patient with Parkinson’s disease who eventually succumbed to metastatic malignant melanoma, secondary to a primary malignant melanoma on the back that was treated 6 years earlier. More research is needed to better characterize the associations between these two conditions, and to evaluate the effectiveness of prevention strategies such as melanoma screening for adults with PD.
|
Views: 1043
HTML: 148
PDF: 506
|
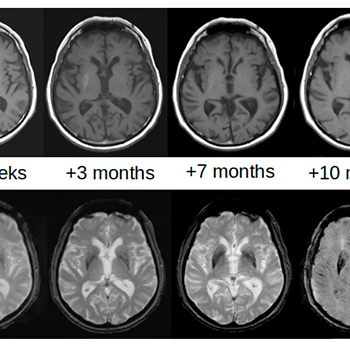
Background: Lateralized involuntary movements consistent with hemichorea-hemiballism (HCHB) may appear following the development of contralateral haemorrhagic or ischaemic lesions of the basal ganglia, particularly the striatum (caudate nucleus and putamen). This condition is called vascular HCHB, but the same symptoms can be caused by a completely different striatal lesion. Glycaemic HCHB may occur in patients with uncontrolled hyperglycaemia: basal ganglia hyperdensity is seen on brain CT, while increased T1 signal intensity and reduced susceptibility-weighted imaging (SWI) and gradient-echo sequences (T2*-GRE) are detected on MRI.
Case description: An 83-year-old man with multiple vascular risk factors and uncontrolled chronic hyperglycaemia was admitted for ischaemic stroke presenting with dysarthria and mild left hemiparesis. No involuntary movements were reported at admission. The emergent brain CT scan was negative for vascular acute lesions, while a mild bilateral hyperdensity of the striata was detectable. Involuntary movements on the left side of the body, consistent with HCHB, appeared 27 days later. The alterations on brain CT completely disappeared after 3 months. On brain MRI, the T1 signal alterations resolved after 10 months, while SWI and T2*-GRE sequences showed persisting alterations after 2 years.
Discussion: Detailed brain imaging demonstrated evolution of striatal alterations of glycaemic HCHB before the appearance of involuntary movements and during the following 2 years. The association between ischaemic stroke and glycaemic HCHB favours the hypothesis that chronic hyperglycaemia more likely determines striatal alterations and the clinical picture of HCHB when vascular hypoperfusion also occurs.
|
Views: 1249
HTML: 188
PDF: 586
|
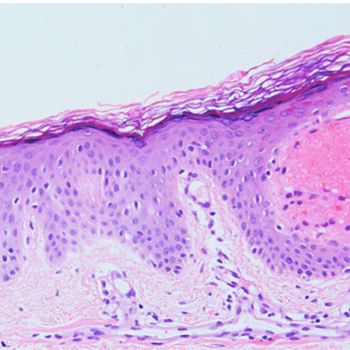
Lysosomal storage disorders (LSDs) are a group of genetic disorders caused by mutations in genes encoding enzymes involved in lysosomal function. Schindler disease is an autosomal recessive, inherited LSD caused by defective or non-existent activity of the enzyme ?-N-acetylgalactosaminidase (?-NAGA). To date, three main phenotypes of Schindler disease have been described. We report the case of a 68-year-old man presenting with axonal and demyelinating polyneuropathy, sensorineural hearing loss, chronic lymphoedema, angiokeratoma corporis diffusum and bilateral carpal tunnel syndrome. Genetic testing (PCR) for ?-galactosidase revealed the c.577G>T (p.Glu193*) mutation in the NAGA gene, confirming Schindler disease, which is clinically compatible with Kanzaki disease and Schindler disease type II.
|
Views: 1065
HTML: 282
PDF: 508
|
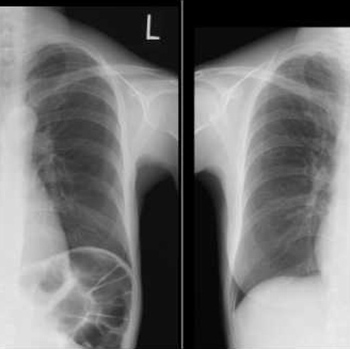
Background: Bronchiolitis obliterans syndrome (BOS) is the term used for the progressive obliteration of small airways before the patient has had a confirmatory lung biopsy. It is also recognized as a transplant-related complication. There have been no reports of BOS during initial standard chemotherapy.
Case presentation: A 50-year-old woman with newly diagnosed follicular lymphoma grade 2, stage 3A, presented with hypoxia and progressive dyspnoea after the fifth cycle of R-CHOP. High-resolution computed tomography showed air trapping enhanced at the end-expiratory phase. Pulmonary function testing revealed severe obstructive and restrictive failure without bronchodilator response. We diagnosed BOS based on current criteria and treated the patient with glucocorticoids and cyclosporin. She was discharged home on oxygen therapy. However, soon after discharge, her respiratory symptoms deteriorated and she was hospitalized in a palliative care unit. She died of respiratory failure within a year of symptom onset.
Conclusions: This is the first case report to describe rapidly progressive BOS in a patient undergoing R-CHOP treatment, which strongly suggests the condition was caused by the chemotherapy. Although a pathological diagnosis was not obtained, the clinical diagnosis of BOS was important so that the patient could receive appropriate treatment and palliative care based on the prognosis of this incurable condition.
|
Views: 1291
HTML: 188
PDF: 582
|
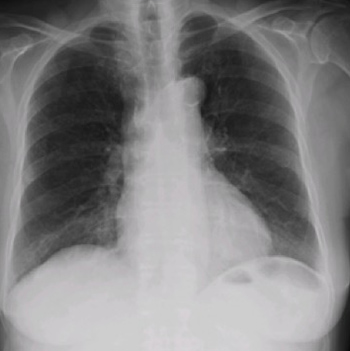
Nitrofurantoin-induced diffuse lung toxicity is well documented in the literature but is often misdiagnosed. We describe an 82-year-old female medicated with nitrofurantoin for the previous 2 years who was admitted for dyspnoea, dry cough and fatigue for 4 months. She was febrile and tachypnoeic and she presented with bilateral basal crackles, hypoxaemic respiratory failure and slightly elevated C-reactive protein levels. A chest radiograph showed bilateral air-space consolidation and interstitial infiltrates and the high-resolution computed tomography scan was evocative of a perilobular pattern of organising pneumonia (OP). Due to the clinical–radiological context, she was diagnosed with a presumable nitrofurantoin-induced OP. She was started on prednisolone 60 mg daily with a progressive improvement. It is important that clinicians are aware of the spectrum of side effects associated with nitrofurantoin so as to monitor patients.
|
Views: 924
HTML: 75
PDF: 500
|
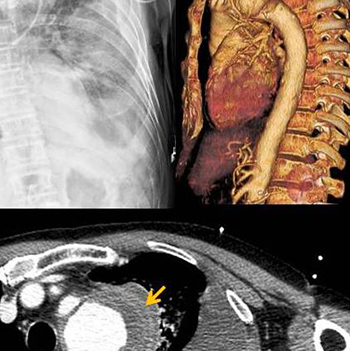
Aortic dissection is a life-threatening clinical emergency and a challenging diagnosis. Depending on its initial location, it may present with several symptoms with the most common being chest pain. We describe the case of a 62-year-old man admitted to the Emergency Department with acute neurological deficits and triaged for the stroke protocol. After unexpected findings on physical examination, other diagnostic hypotheses were evaluated, culminating in the diagnosis of aortic dissection with haemothorax mimicking a stroke.
|
Views: 1047
HTML: 98
PDF: 482
|
Pulmonary arteriovenous malformations (PAVMs) are rare vascular anomalies. Alternative designations are pulmonary arteriovenous fistulae or aneurysms. Although mostly asymptomatic, PAVMs can cause respiratory symptoms due to right-to-left shunt. The central nervous system is a potential target for complications, including stroke, as a result of paradoxical embolism. In this report, the authors describe an unusual case of cerebral emboli caused by paradoxical embolism through a PAVM, presenting with a broad pathology including orthodeoxia, central cyanosis and digital clubbing, which should be kept in mind since misdiagnosis may cause severe morbidity in young adults.
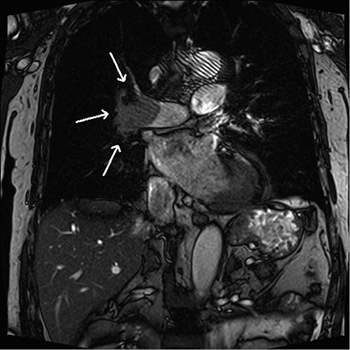
|
Views: 939
HTML: 110
PDF: 354
|
In this case report we describe a 69-year-old male ex-smoker with non-small-cell carcinoma localised within the pulmonary artery, misdiagnosed as pulmonary thromboembolism. This case indicates that non-small-cell carcinoma can be localised within the pulmonary artery. Furthermore, it emphasises the importance of performing a positron emission tomography scan and diagnostic intraluminal biopsy in unexplained lesions in the pulmonary artery to reach the proper diagnosis at the early stages of the disease. The patient is currently undergoing concurrent chemotherapy and radiation therapy.
|
Views: 1173
HTML: 120
PDF: 546
|
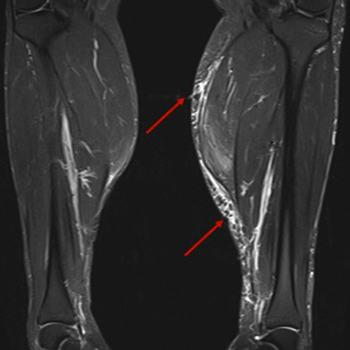
A 43-year-old Caucasian male initiated myalgias and loss of muscle strength in the upper and lower limbs, but especially at the shoulder and pelvic girdle. Creatinine phosphokinase was elevated seven-fold above the normal reference value and aldolase was slightly elevated. He had a previous diagnosis of Behçet's disease, antiphospholipid syndrome and hypertriglyceridaemia. At this time, he was on azathioprine 150 mg daily, colchicine 1 mg daily, warfarin and fenofibrate 200 mg daily. Fenofibrate was stopped and creatinine phosphokinase re-evaluated 2 months later, but it was higher, with persistent myalgias. By this time, prednisolone was restarted and the azathioprine dose reduced until it was discontinued. Nevertheless, 2 months after stopping azathioprine, the patient remained symptomatic and creatinine phosphokinase was persistently elevated. At this point, the authors requested myositis antibody testing to exclude overlap with a third autoimmune disorder, and Ro52 antibody was positive. Electromyography was normal. Magnetic resonance imaging of lower limb muscles was compatible with polymyositis. Muscular biopsy of the medial gastrocnemius revealed inflammatory myopathy. The authors proposed treatment with rituximab and after 3 months, the patient had clinically and analytically improved, with reduction of creatinine phosphokinase, without adverse reactions. As we can see in this case, rituximab could be a secure treatment for patients with idiopathic inflammatory myopathy without improvement on glucocorticoids plus another immunosuppressive agent. This patient has a rare overlap syndrome, since this is the first case of an association between inflammatory myopathy, Behçet's disease and antiphospholipid syndrome described in the literature.
|
Views: 1140
HTML: 571
PDF: 433
|
Systemic lupus erythematosus (SLE) is a chronic multi-systemic immune-mediated disease with confusing symptoms and delayed diagnosis. We report the case of a 32-year-old man with a persistent Venereal Disease Research Laboratory (VDRL)-positive reaction treated for syphilis 5 years previously, who was admitted for rash, weight loss, pancytopenia, inflammatory syndrome, and an important spontaneous prolongation of activated partial thromboplastin time (aPTT). Antiphospholipid antibodies were identified in the patient and he was diagnosed with SLE. The unrecognized false positive VDRL reaction and the delayed diagnosis of SLE were harmful as the patient had developed renal and cardiac complications by the time of diagnosis.
|
Views: 1629
HTML: 379
PDF: 1159
|
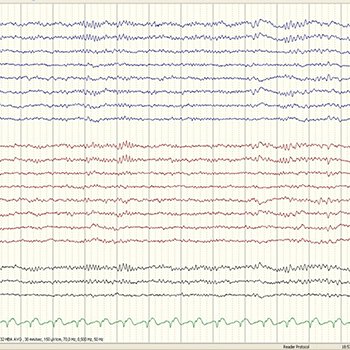
Spindle coma is an electroclinical entity that has been used to describe an EEG pattern of “sleep-like” activity in comatose patients. Although it has been associated with favourable prognosis, its aetiology is one of the key factors for patient outcome. The authors present three cases of spindle coma with different aetiologies (amitriptyline overdose, pontine myelinolysis and hypoxic-ischaemic encephalopathy) that culminated in different outcomes.
|
Views: 1195
HTML: 138
PDF: 497
|
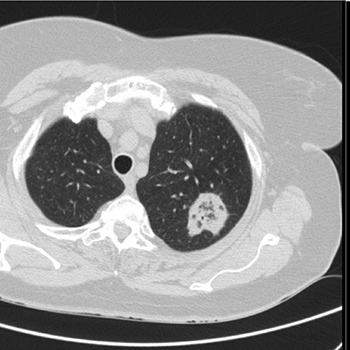
The reversed halo sign is defined as a focal rounded area of ground-glass opacity surrounded by a more or less complete ring of consolidation. It is a relatively rare sign and initially considered a specific sign of organising pneumonia. We report the case of a 55-year-old female who was being followed-up in a pulmonology consultation due to a 6 mm nodule which required vigilance. On a re-evaluation chest CT scan, besides a stable 6 mm nodule, a 36 mm mass with the reversed halo sign was diagnosed. The presence of the reversed halo sign misled the multidisciplinary team into the diagnosis of organising pneumonia and initiation of corticotherapy was suggested. However, after further investigation, a final diagnosis of pulmonary tuberculosis was made. Even though this sign is relatively rare, and still considered an important clue to the diagnosis of organising pneumonia in immunocompetent patients, other causes must be excluded before starting treatment.
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2024, Published by SMC Media srl, Italy - Privacy policy

