EJCRIM 2023 CiteScore
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |
Last updated on 05 May, 2024
Updated monthly
Updated monthly
Powered by


|
Views: 1150
HTML: 173
PDF: 626
|
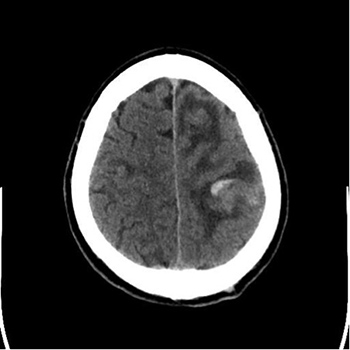
Varicella infection is caused by varicella-zoster virus (VZV) and commonly presents as a self-limiting skin manifestation in children. VZV also causes cerebral arterial vasculopathy and antibody-mediated hypercoagulable states leading to thrombotic complications in children, although there are very few such reports in adults. Postulated causal factors include vasculitis, direct endothelial damage, or acquired protein S deficiency secondary to molecular mimicry. These induced autoantibodies to protein S could lead to acquired protein S deficiency and produce a hypercoagulable state causing venous sinus thrombosis. Here we report the case of a 26-year-old man who presented with cortical venous sinus thrombosis and acute pulmonary embolism following varicella infection. Both conditions responded to anticoagulation treatment.
|
Views: 1309
HTML: 100
PDF: 605
|
Background: Antiphospholipid syndrome (APS) is defined as thromboembolic complications and/or pregnancy morbidity in the presence of persistent increased titres of antiphospholipid antibodies. Nevertheless, some patients with clinical signs suggestive of APS are negative for diagnostic antibodies and may be classified as having seronegative-APS (SN-APS). Among the ‘non-diagnostic’ antibodies, a few studies have suggested that the IgG anti-vimentin/cardiolipin antibodies (AVA/CL) may be associated with risk of thrombosis.
Aims: The aim of this case report is to encourage the assessment of non-conventional antibodies in APS.
Patient and methods: We report the case of a 69-year-old male patient with rapid onset of apparently unexplained multiple exclusively arterial thrombotic events in both coronary and peripheral vascular beds.
Results: The patient did not meet the diagnostic criteria for APS but was positive for AVA/CL, which result persisted on further testing at 3 and 6 months.
Discussion: Ongoing research has revealed the existence of non-criteria antibodies which may be relevant for the diagnosis of SN-APS and should be included in the classification criteria for the disease.
|
Views: 1594
HTML: 206
PDF: 577
|
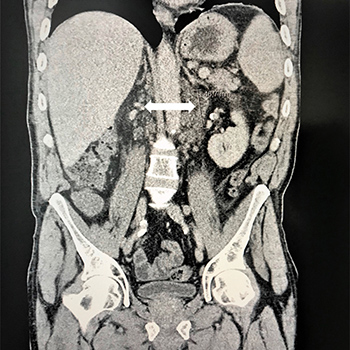
Whipple's disease is a rare multisystemic infectious disease that can mimic lymphoproliferative disorders and must be considered in the differential diagnosis of febrile syndromes. The authors describe the case of a 55-year-old man who presented to the Emergency Department with dyspnoea and abdominal pain. He had a 2-month history of fever, night sweats, asthenia and unintentional weight loss. Upon clinical examination he had bilateral inguinal lymphadenopathy. Blood tests showed iron-deficit anaemia and high C-reactive protein. Abdominal ultrasound showed mesenteric and iliac adenopathies and hepatosplenomegaly. The patient was admitted to the Internal Medicine department for additional testing. Flow cytometry analysis of peripheral blood showed CD5-positive monoclonal B-cell expansion. Excisional biopsy of a retroperitoneal adenopathy guided by computed tomography showed periodic acid–Schiff-positive bacilli inside the macrophages, further identified as Tropheryma whipplei through polymerase chain reaction. Bone marrow biopsy showed a scarce positive CD5 lymphoid population and haematopoietic alterations related to infection. The patient started treatment for T. whipplei with complete symptom resolution. This is the first case describing the simultaneous diagnosis of Whipple's disease and chronic lymphocytic leukaemia in a patient with constitutional symptoms, fever and lymphadenopathies.
|
Views: 874
HTML: 108
PDF: 496
|
Acquired causes of Fanconi syndrome in adults are usually due to drugs, toxins or paraproteinaemias. Infectious causes are rarely described. We report a case of invasive pneumococcal disease in a patient who developed a Fanconi-like syndrome during the course of her illness. This patient presented with multiple electrolyte derangements consisting predominantly of hypokalaemia, hypomagnesaemia and hypophosphataemia during hospitalization for invasive pneumococcal disease with possible Austrian syndrome. Further evaluation revealed significant urinary losses of these electrolytes, uric acid and ?2-microglobulin. Together with evidence of hypouricaemia, this is suggestive of proximal renal tubulopathy, and hence a Fanconi-like syndrome. The patient’s clinical condition and biochemical anomalies improved following pneumococcus treatment.
|
Views: 1094
HTML: 267
PDF: 517
|
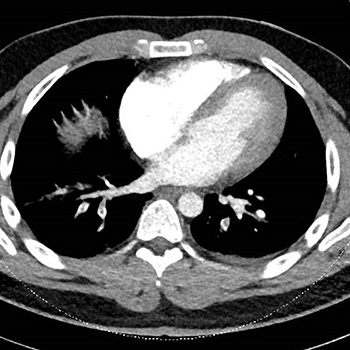
Spontaneous haemothorax complicating the treatment of pulmonary embolism is rare and potentially fatal. We describe a patient with pulmonary embolism and severe pleuritic pain who developed a life-threatening haemothorax 10 days later while on rivaroxaban therapy. This case highlights the fact that severe pleuritic pain associated with pulmonary embolism may indicate subclinical infarction of tissue near the visceral pleura with an increased risk of pleural effusion and the subsequent development of a haemothorax. It is important to recognise such danger signs warranting closer attention, especially since the increased use of direct oral anticoagulants has facilitated ambulatory care and this complication may manifest in the outpatient setting.
|
Views: 896
HTML: 159
PDF: 469
|
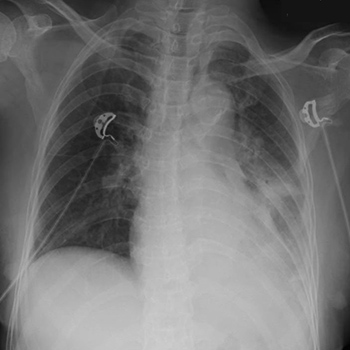
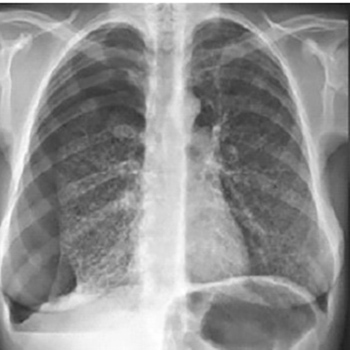
Tuberous sclerosis complex (TSC) is a rare, autosomal dominant disorder with a recognized phenotypic variability with clinical manifestations developing continuously throughout life. The follow-up of TSC patients is challenging. The authors present a case with a definite diagnosis of TSC with known skin, renal, hepatic and neuropsychiatric involvement, whose diagnosis of TSC-associated lymphangioleiomyomatosis was establish at a late stage after the patient had presented with recurrent pneumothorax. Notwithstanding, mammalian target of rapamycin inhibition therapy was initiated.
|
Views: 936
HTML: 104
PDF: 618
|
A 32-year-old woman undergoing an in vitro fertilization program was admitted to our hospital with the diagnosis of severe ovarian hyperstimulation syndrome (OHSS). Transvaginal ultrasonography showed two gestational sacs. Treatment with fluid restriction, serum albumin and intravenous furosemide was started, and repeated thoracentesis and paracenteses were performed. In the absence of clinical improvement, the patient was transferred to the intensive care unit and a therapeutic abortion was suggested. Due to the similarities between OHSS and idiopathic systemic capillary leak syndrome, we offered the patient compassionate treatment with intravenous immunoglobulins.
After administration, the patient showed rapid improvement and we were able to suspend intravenous furosemide and serum albumin. She was discharged, and pregnancy has continued normally to date.
|
Views: 1397
HTML: 176
PDF: 465
|
Boerhaave syndrome or spontaneous rupture of the oesophagus is a severe condition commonly misdiagnosed or unrecognized. Prognosis is poor even if the diagnosis is made promptly. We describe a case of Boerhaave syndrome diagnosed after the development of pneumomediastinum and cardiac arrest. Unfortunately, the patient died 48 hours after admission to the Intensive Care Unit. This entity requires a multidisciplinary management approach which may include conservative, surgical or endoscopic procedures.
|
Views: 1530
HTML: 195
PDF: 604
|
We report the case of a 28-year-old man who presented with recurring episodes of high fever, pleural and pericardial effusions and bilateral hydrocele. He was diagnosed with familial Mediterranean fever (FMF) and responded well to colchicine therapy. Genetic testing showed variants of the MEFV gene (p.Pro369Ser and p.Glu148Gln) previously independently described as having a more benign course of the disease. Their association is very rarely reported. Our patient and our review of the literature suggest that these genetic variants are associated with indolent courses but might also trigger the classic symptoms seen in severe FMF, probably in a compound heterozygous fashion. The combination of these variants should be taken into consideration in the diagnosis and management of patients.
|
Views: 19872
HTML: 4574
PDF: 34171
|
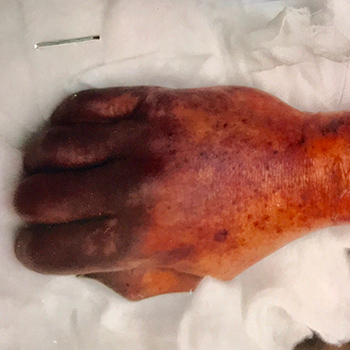
Bite infections caused by Capnocytophaga canimorsus are rare. Severe and fatal infections are more frequently reported in patients with immunodeficiency, splenectomy or alcohol abuse. We describe the case of a 63-year-old man who developed flu-like symptoms and presented after some delay with severe sepsis and purpura fulminans. He was found to be infected with C. canimorsus without a bite injury and did not demonstrate immunodeficiency or any other typical predisposition. Despite extensive intensive care, his conditions deteriorated and he died from multiorgan failure.
|
Views: 1222
HTML: 83
PDF: 695
|
Hyponatraemia is a common electrolyte abnormality seen by internists. Clinical features of hyponatraemia are primarily related to CNS dysfunction, and depend on the severity and acuity of changes in serum sodium concentration. Neurological manifestations of hyponatraemia range from nausea and malaise, with a mild reduction in the serum sodium, to lethargy, decreased level of consciousness, headache, seizures and coma in extreme cases. Focal neurological deficits are very rare in the setting of hyponatraemia. Here, we describe a patient with acute severe symptomatic hyponatraemia presenting with focal neurological deficits that resolved after correction of acute hyponatraemia.
|
Views: 1620
HTML: 1011
PDF: 701
|
Introduction: Osteonecrosis of the jaw has been consistently reported in the literature associated to the high-dose intravenous bisphosphonate therapy. However, osteonecrosis can also occur in patients who have other risk factors.
Case description: An unusual case of ONJ in a patient being treated with esomeprazole is reported.
Discussion: The probable association between proton pump inhibitor intake and osteonecrosis of the jaw should alert clinicians. Collaborations between medical and dental doctor and an early diagnosis might prevent or reduce the morbidity resulting from advanced destructive lesions of the jaw bone.
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2024, Published by SMC Media srl, Italy - Privacy policy

